Theoretical Investigations on the Mechanisms for the Reactions of Sevoflurane Radicals[(CF3)2C(·)OCH2F, (CF3)2CHOC(·)HF] with O2 and the OH· Radicals Regeneration
Received Date:
04 August 2018 Available Online:
15 October 2018
Fund Project:
Project supported by the National Key Research and Development Program of China (No. 2017YFB0902500) and Science and Technology Project of State Grid Corporation of China: The key Technology of Environment-Friendly Gas-Insulated Transmission Line (GIL)
Abstract:
Sevoflurane is an excellent volatile anaesthetic which has been widely in clinical use. However, it was found that sevoflurane is a potent green-house gas with a significant global warming potential. Atmospheric degradation of sevoflurane is desired for its long-term application. The reaction of sevoflurane with hydroxyl radicals (OH·) produces two radical species, namely, (CF3)2C(·)OCH2F and (CF3)2CHOC(·)HF, which have different reactivity. Under the low-NO atmospheric conditions, it was found that both radical fragments enable to initialize the regeneration of OH·radicals in the presence of molecular oxygen (O2). Microscopic mechanisms for the reactions of the two radicals with O2 have been investigated for the first time in this work. Geometries of various intermediates and transition states on the doublet potential energy surfaces were optimized at the M06-2X/6-311++G (d, p) level of theory. Moreover, the single-point calculations were carried out using the composite model CBS-Q to refine the reaction energetics to the chemical accuracy. It was revealed that the formation of peroxy intermediate (RO2·) undergoes via the definitive barriers of 1.3 or 1.8 kcal·mol-1, in contrast to the barrierless association between the alkyl radicals and O2. Apparently, the association of the fluorinated alkyl radicals with O2 takes place more slowly due to the substitute effect. Although the addition of O2 to the fluorine-rich radical site is more preferable than that to the fluorine-poor site, the latter is more exothermic in view of the exothermicity of the intermediates RO2·. The barriers for the subsequent H-migration of RO2·to form the QOOH intermediates are 17.9 and 21.5 kcal·mol-1, respectively. Both barriers lie well below the reactant asymptote, indicating the isomerization paths are energetically favorable. Decomposition of QOOH takes place via three competitive mechanisms, including the step-wise bond fission, the three-body concerted cleavage, and the four-center intramolecular SN2 reaction, to produce OH·radicals predominantly. All the reaction pathways could be competitive for (CF3)2C(OC(·)HF)OOH because the energies of the corresponding barriers are close. In contrast, only the SN2 displacement energetic route is dominant for (CF3)2C(·)OC(HF)(OOH). Neither step-wise nor three-body pathways is important because the barrier height is roughly 7 kcal·mol-1 higher than that for the SN2 pathway. The isomerization of QOOH to alkoxy intermediate is of little importance due to the significant barrier even though it is highly exothermic. Implication of the current theoretical findings in the OH·radicals recycling reaction in atmosphere has been illustrated.
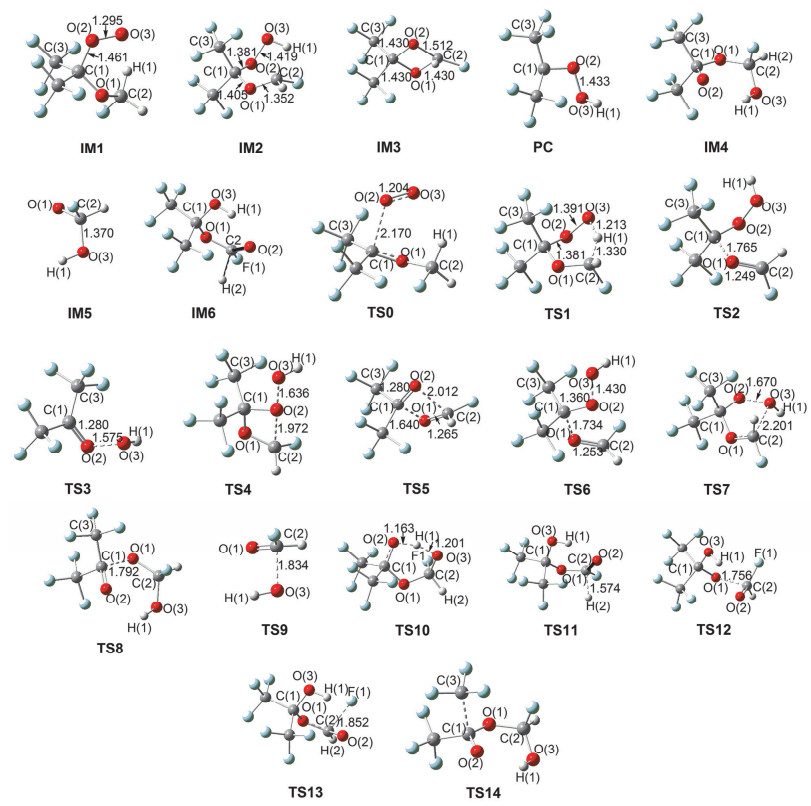
Figure 2.
M06-2X/6-311++G(d, p) optimized geometrical parameters for the species involved in the (CF3)2C(•)OCH2F+O2 reaction. Bond distances are in ngstroms
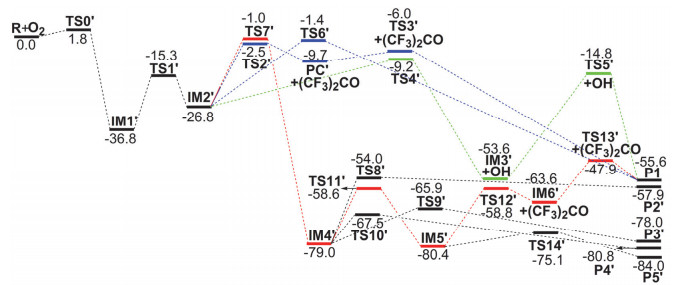
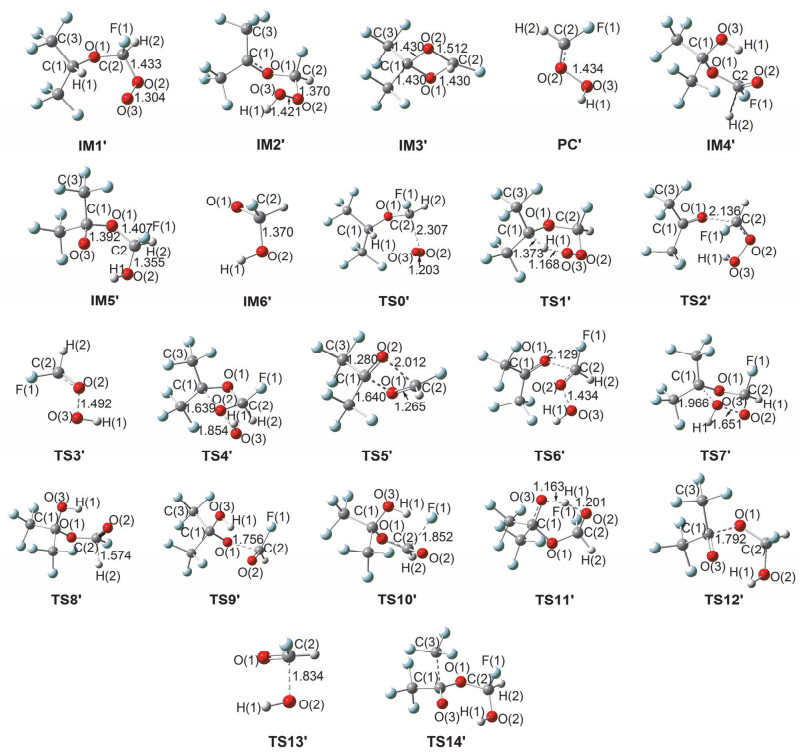
Figure 4.
M06-2X/6-311++G (d, p) optimized geometrical parameters for the species involved in the (CF3)2CHOC(•)HF+O2 reaction. Bond distances are in ngstroms
Sulbaek Andersen, M. P.; Sander, S. P.; Nielsen, O. J.; Wagner, D. S.; Sanford, T. J.; Wallington, T. J. Brit. J. Anaesth. 2010, 105(6), 760. doi: 10.1093/bja/aeq259
[5]
Brown, A. C.; Canosa-Mas, C. E.; Parr, A. D.; Wayne, R. P. Atmos. Environ. 1990, 24(9), 2499. doi: 10.1016/0960-1686(90)90341-J
Wilson, Jr. E. W.; Hamilton, W. A.; Mount, H. R. J. Phys. Chem. A 2007, 111(9), 1610. doi: 10.1021/jp068355d
[8]
Vollmer, M. K.; Rhee, T. S.; Rigby, M.; Hofstetter, D.; Hill, M.; Schoenenberger, F.; Reimann, S. Geophys. Res. Lett. 2015, 42, 1606. doi: 10.1002/2014GL062785
[9]
Brown, A. C.; Canosa-Mas, C. E.; Parr, A. D.; Pierce, J. M. T.; Wayne, R. P. Nature 1989, 341, 635. doi: 10.1038/341635a0
[10]
Langbein, T.; Sonntag, H.; Trapp, D.; Hoffmann, A.; Malms, W.; Röth, E. P.; Mörs, V.; Zellner, R. Brit. J. Anaesth. 1999, 82(1), 66. doi: 10.1093/bja/82.1.66
[11]
Sulbaek Andersen, M. P.; Nielsen, O. J.; Wallington, T. J.; Karpichev, B.; Sander, S. P. Anesth. Analg. 2012, 114, 1081. doi: 10.1213/ANE.0b013e31824d6150
[12]
Mishra, B. K.; Lily, M.; Chakrabartty, A. K.; Bhattacharjee, D.; Deka, R. C.; Chandra, A. K. New J. Chem. 2014, 38, 2813. doi: 10.1039/C3NJ01408H
Walker, R. W. In Research in Chemical Kinetics, Vol. 3, Eds. : Compton, R. G. ; Hancock, G., Elsevier, Amsterdam, The Netherland, 1995, p. 1.
[18]
Walker, R. W. ; Morley, C. In Low-Temperature Combustion and Autoignition, Ed. : Pilling, M. J., Elsevier, Amsterdam, The Netherland, 1997, pp. 1~124.
[19]
Robertson, S. H. ; Seakins, P. W. ; Pilling, M. J. In Low-Temperature Combustion and Autoignition, Ed. : Pilling, M. J., Elsevier, Amsterdam, The Netherland, 1997, p. 125.
[20]
Pollard, R. T. In Gas Phase Combustion, Eds. : Bamford, C. H. ; Tipper, C. F. H., Elsevier, New York, 1977, p. 249.
[21]
DeSain, J. D.; Taatjes, C. A.; Miller, J. A.; Klippenstein, S. J.; Hahn, D. K. Faraday Discuss. 2001, 119, 101. doi: 10.1039/b102237g
[22]
Eskola, A. J.; Carr, S. A.; Shannon, R. J.; Wang, B.; Blitz, M. A.; Pilling, M. J.; Seakins, P. W. J. Phys. Chem. A 2014, 118, 6773. doi: 10.1021/jp505422e
Zhao, Y.; Truhlar, D. G. Acc. Chem. Res. 2008, 41(2), 157. doi: 10.1021/ar700111a
[25]
Gonzalez, C.; Schlegel, H. B. J. Chem. Phys. 1989, 90, 2154. doi: 10.1063/1.456010
[26]
Montgomery, J. A.; Frisch, M. J.; Ochterski, J. W.; Petersson, G. A. J. Chem. Phys. 1999, 110, 2822. doi: 10.1063/1.477924
[27]
Frisch, M. J. ; Trucks, G. W. ; Schlegel, H. B. ; Scuseria, G. E. ; Robb, M. A. ; Cheeseman, J. R. ; Scalmani, G. ; Barone, V. ; Mennucci, B. ; Petersson, G. A. ; Nakatsuji, H. ; Caricato, M. ; Li, X. ; Hratchian, H. P. ; Izmaylov, A. F. ; Bloino, J. ; Zheng, G. ; Sonnenberg, J. L. ; Hada, M. ; Ehara, M. ; Toyota, K. ; Fukuda, R. ; Hasegawa, J. ; Ishida, M. ; Nakajima, T. ; Honda, Y. ; Kitao, O. ; Nakai, H. ; Vreven, T. ; Montgomery Jr., J. A. ; Peralta, J. E. ; Ogliaro, F. ; Bearpark, M. ; Heyd, J. J. ; Brothers, E. ; Kudin, K. N. ; Staroverov, V. N. ; Kobayashi, R. ; Normand, J. ; Raghavachari, K. ; Rendell, A. ; Burant, J. C. ; Iyengar, S. S. ; Tomasi, J. ; Cossi, M. ; Rega, N. ; Millam, J. M. ; Klene, M. ; Knox, J. E. ; Cross, J. B. ; Bakken, V. ; Adamo, C. ; Jaramillo, J. ; Gomperts, R. ; Stratmann, R. E. ; Yazyev, O. ; Austin, A. J. ; Cammi, R. ; Pomelli, C. ; Ochterski, J. W. ; Martin, R. L. ; Morokuma, K. ; Zakrzewski, V. G. ; Voth, G. A. ; Salvador, P. ; Dannenberg, J. J. ; Dapprich, S. ; Daniels, A. D. ; Farkas, O. ; Foresman, J. B. ; Ortiz, J. V. ; Cioslowski, J. ; Fox, D. J. Gaussian 09, Revision A. 02, Gaussian, Inc., Wallingford, CT, 2009.
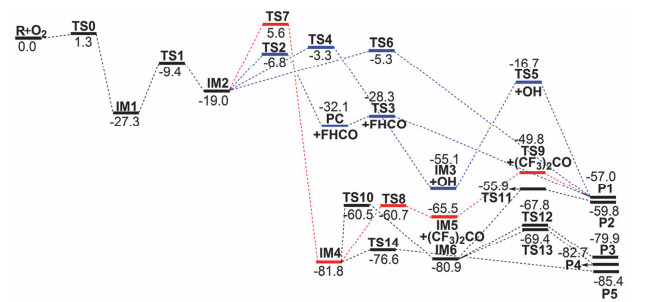
图 1
在CBS-Q理论水平下(CF3)2C(•)OCH2F+O2反应的势能剖面示意图
Figure 1
Energetic profiles (in kcal•mol-1) for the (CF3)2C(•)OCH2F+O2 reaction at the CBS-Q level of theory
Figure 2
M06-2X/6-311++G(d, p) optimized geometrical parameters for the species involved in the (CF3)2C(•)OCH2F+O2 reaction. Bond distances are in ngstroms
Figure 4
M06-2X/6-311++G (d, p) optimized geometrical parameters for the species involved in the (CF3)2CHOC(•)HF+O2 reaction. Bond distances are in ngstroms

 下载:
下载:



 下载:
下载:





 下载:
下载:
