Received Date:
10 July 2019 Revised Date:
22 December 2019 Available Online:
10 April 2020
Abstract:
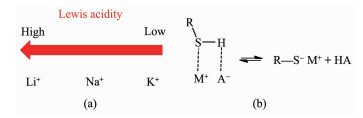
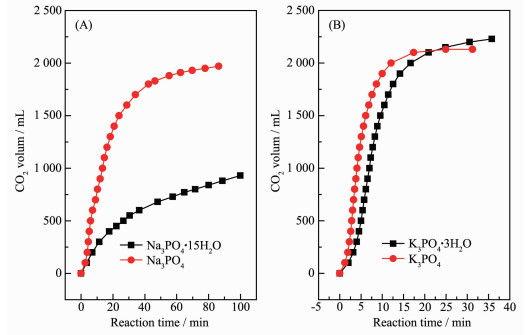
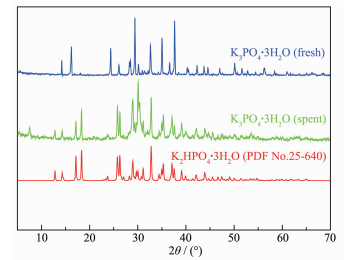
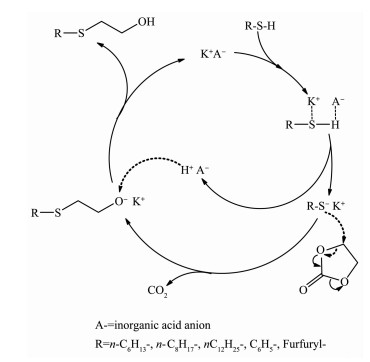
For the β-hydroxyethylation of sulfur atoms in thiols, there should be a strong interaction between the -SH group and the methylene carbon of ethylene carbonate (EC). However, the nucleophilicity of -SH group is insufficient to interact with the methylene carbon of EC. Therefore, a catalyst should be required to activate -SH group. Under the conditions of atmospheric pressure, solvent-free and 120℃, 14 kinds of alkali metal mineral acid salts (MA, M=Li+, Na+ and K+) were investigated for the S-hydroxyethylation of n-octyl mercaptan with EC. When A- of MA are the same, the ionic radius and polarizability of Li+, Na+ and K+ increase sequentially, the ability of MA to activate SH group also increase sequentially. The general rule is that the catalytic activity of the potassium salt is higher than that of the corresponding sodium salt, while the lithium salt has no catalytic activity. In various inorganic acid potassium salts, the catalytic activity of KA has a positive correlation with the pKa of its corresponding conjugate acid HA. The catalytic activity of KA is lower when its conjugate acid HA is more acidic. In order to explore the universality of the mechanism of potassium salt catalytic activation of -SH, K3PO4 was used as catalyst to investigate the catalytic activity of β-hydroxyethylation of six different structures of thiols with EC at different reaction temperatures. Due to the acidic difference of mercaptan, the rule is that the stronger the acidity of the mercaptan, the higher the reactivity, and the easier the S-H bond is to dissociate. The advantages of the reaction are as follows:no solvent is involved, the molar ratio of mercaptan to EC is close to the theoretical amount, the selectivity of the product β-hydroxyethyl sulfide is >99%, no halogen containing reactant involved and therefore no halogen salt byproduct is formed in the final product.
a Reactions were carried out with n-C8H17SH (110 mmol), molar ratio of MA to n-C8H17SH is 1/100 and molar ratio of EC to n-C8H17SH is 1.02 under atmospheric pressure at 120 ℃; b Conjugated acid of MA, pKa1 data are from reference[26]; c Determined by gas chromatography (GC); d molar ratio of KHCO3 to n-C8H17SH is 1/50.
a Reactions were carried out with RSH (110 mmol), molar ratio of K3PO4 to RSH is 1/100 and molar ratio of EC to RSH is 1.02 under atmospheric pressure.
Table 1.
Activity of alkali metal mineral acid salts (MA) catalyzed β-hydroxyethylation of n-octyl mercaptan with ECa
Entry
MA
pKa1 of HAb
Reaction time / min
CO2 volume / mL
Con. / %c
Sel. / %
1
None
—
120
0
0
0
2
Li3PO4
2.16
120
0
0
0
3
Na3PO4
86.3
1 970
87.3
95.2
4
K3PO4
25
2 130
97.4
100
5
Na3PO4·12H2O
99
930
37.7
100
6
K3PO4·3H2O
71.8
2 340
98.8
100
7
KH2PO4
120
0
0
0
8
K2HPO4·3H2O
120
0
0
0
9
K4P2O7
0.91
193
1 570
66.7
100
10
Li2CO3
6.38
180
0
0
0
11
Na2CO3
204.45
1 640
68.7
100
12
K2CO3
41.53
2 240
98.6
100
13
KHCO3d
51.2
2 170
95.5
100
14
Na2B4O7·10H2O
4.00
188
830
34.0
100
15
K2B4O7·4H2O
74
2 160
90.5
98.7
16
Na2MoO4·2H2O
2.54
126
1 850
93.4
99.3
17
K2MoO4
96
2 020
96.4
99.4
18
Na2SnO3·3H2O
2.57
270
940
39.2
99.9
19
K2SnO3·3H2O
84.1
2 135
94.9
99.6
20
Na2SiO3
9.9
265.7
1 860
76.3
100
21
K2SiO3
122.5
2 040
83.2
100
22
Na2SO4
-3.0
120
0
0
0
23
K2SO4
101.5
800
34.2
100
24
NaF
3.15
120
0
—
—
25
KF·2H2O
167
1 870
94.7
99.9
26
KF
162
2 090
95.4
98.2
27
NaCl
-8.00
176.5
1 770
89.1
97.3
28
KCl
126.38
2 100
92.4
97.6
29
NaBr
-9.00
340.15
800
75.4
100
30
KBr
128.55
1 500
76.9
99.4
a Reactions were carried out with n-C8H17SH (110 mmol), molar ratio of MA to n-C8H17SH is 1/100 and molar ratio of EC to n-C8H17SH is 1.02 under atmospheric pressure at 120 ℃; b Conjugated acid of MA, pKa1 data are from reference[26]; c Determined by gas chromatography (GC); d molar ratio of KHCO3 to n-C8H17SH is 1/50.
Table 2.β-hydroxyethylation of various thiol compounds with EC catalyzed by K3PO4
R
Temperature / ℃
Time / min
CO2 volume / mL
RSH Con. / %
Sel. / %
n-C6H13-
120
41.9
2 240
92.4
100
110
44.2
2 140
91.9
100
n-C8H17-
120
24.9
2 130
91.4
100
110
82.4
2 140
91.2
100
n-C12H25-
120
55.5
2 185
93.8
100
C6H5-
90
15.18
2 460
99.9
100
70
42.33
2 400
99.6
100
HOCH2CH2-
120
15.0
2 450
99.4
99.4
100
15.2
2 270
92.9
99.6
Furfuryl-
100
28.5
2 455
99.7
100
80
61.2
2 275
92.4
100
a Reactions were carried out with RSH (110 mmol), molar ratio of K3PO4 to RSH is 1/100 and molar ratio of EC to RSH is 1.02 under atmospheric pressure.

 下载:
下载:

 下载:
下载:





 下载:
下载:
