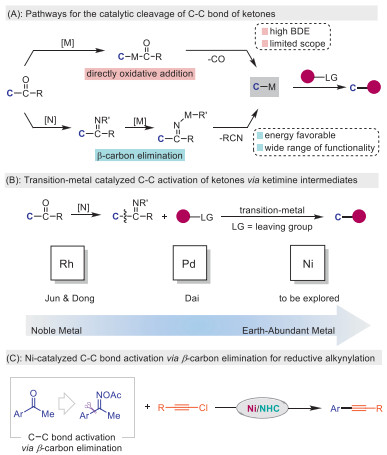
Scheme 1.
Transition metal-catalyzed unstrained C–C bond activation for cross coupling.
Nickel-catalyzed reductive alkynylation of ketoimines via unstrained C–C bond activation
Tian-Zhang Wang , Le-Yu Tang , Yu-Qiu Guan , Lingfei Hu , Gang Lu , Yu-Feng Liang

Carbon–carbon bond is one of the most prevalent chemical bonds in organic molecules, and it is a very complicated task to cleave and functionalize them simply and efficiently [1–4]. However, there are further problems in this sector related to kinetics and thermodynamics: (1) its chemical bonds are very stable, and the bond dissociation energy is about 90 kcal/mol, making it thermodynamically steady; (2) the C–C bond has a "hidden" nature due to its orbital direction and steric hindrance are constrained, so it is widely distributed throughout the molecular structure [5,6]. Therefore, a great majority of the carbon–carbon bonds in organic compounds exhibit remarkable inertness. There have been many reports of cleavage of the carbon–carbon bonds of small rings, driven by releasing ring strain [7–10]. The development of transition-metal catalysts has become a useful tool for devising unusual bond-disconnecting strategies, especially direct activation of non-strained carbon–carbon bonds, which has also been explored [11–14]. The selective activation of carbon–carbon bonds have the potential to swiftly enhance the complexity of molecules, promising significant opportunities for advancements in materials, medicine, and biology [15–18].
Ketones are a widely present and unstrained chemical motif, with their transformations mainly concentrated in the addition reaction of C=O bond and the activation of C–H bond [19,20]. However, the high bond energy and strong metal affinity of the C=O bond in ketones make it challenging to directly utilize ketones for C–C(O) bond activation [21,22]. Nevertheless, due to the planar structure of ketones, they exhibit lower energies, enabling the activation of C–C bonds associated with carbonyl groups [23]. Therefore, the exploration of C–C(O) bonds using transition metal-catalyzed ketones is a crucial research area that can provide new insights into carbonyl group conversions in the future. In terms of reaction mechanism, it mainly focuses on two reaction modes: (1) the directly metal-catalyzed oxidative addition into the C–C(O) bond, (2) the organometallic intermediates obtained through β-carbon elimination (Scheme 1A). With the strategy of C–C bond activation via oxidative addition, Shi reported the decarbonylation of rhodium-catalyzed directly activating the C–C(O) bond of diaryl ketones successively [24,25]. Subsequently, Dong observed the Rh-catalyzed decarbonylation of ynones [26,27]. As a representative of Earth-abundant metal, nickel catalysts have also shown good effects on the activation of carbon–carbon bonds in recent years. The decarbonylation of diaryl ketones with nickel catalysts has been studied intensively by Martin [28], Chatain [29], Wei [30], and Shi [31]. These reports mainly focus on the direct oxidative addition of the C–C(O) bond of the ketone by the metal catalyst. Whereas this method requires an amount of additional energy and imposes specific activity requirements on substrates due to their high steric hindrance and entropy.
Alternatively, the C–C bond activation via β-carbon elimination process, which is regarded as the inverse reaction of the carbon-metal migratory insertion into double bonds, is not well-developed because the resulting intermediates form a weak carbon-metal bond instead of a stable C–C σ-bond [32,33]. The conversion of ketones into oxime esters is a very straightforward process, and the more polarized N–O bonds in ketoimines can be inserted more easily by transition-metal catalysts [34–36]. This allows the cleavage of C–C bonds through β-carbon elimination for diverse valuable transformations [37–40]. Through this lower barrier approach, Jun developed rhodium-catalyzed C–C bond activation of unstrained imine [41]. Recently, Dong et al. utilized rhodium catalyst and 2-aminopyridine as a co-catalyst to form an imine temporary directing group, facilitating the activation of the C–C bond in the ketone substrate [42,43]. Dai et al. disclosed the transformation of arylketone oxime ester as an arylation reagent catalyzed by palladium [44–46]. Definitely, these strategies have the potential to effectively lower the reaction energy barrier, resulting in milder reaction conditions and a broader substrate range (Scheme 1B). Despite their significance, the capability to achieve selective unstrained C–C bond activation via β-carbon elimination with earth-abundant nickel catalysis remains a longstanding challenge.
Cross electrophile coupling is a powerful strategy in organic synthesis and has been widely used in the last decades [47–50]. Inspired by the elegant contributions [51–91], we envisioned that the nickel catalyst may act as a bifunctional catalyst for the activation of the C–C bond via β-carbon elimination and followed cross coupling with electrophiles (Scheme 1C). However, the challenges of this anticipated reductive transformation are: (1) competition of C–N coupling vs. β-carbon elimination of ketimine intermediates; (2) inhibition of homocoupling of electrophiles; (3) chemo-selectivity issues. Herein, we wish to report the nickel-catalyzed reductive cross-coupling of ketoimines with alkynyl halides via unstrained C–C bond activation. The merits of this method are 3-fold: (1) aryl ketones could participate in the cross electrophile coupling as aryl electrophiles through Ni-catalyzed β-carbon elimination, which will broaden the aryl electrophiles source of the reductive coupling reaction and enrich the diversity of reaction types; (2) the reaction system is simple and eliminate the need of any strong bases or acids, leading to excellent functional group compatibility and successful application in late-stage functionalization of complex molecules; (3) the unique chemo-selectivity of nickel/NHC system in cross-coupling reactions with halides has been verified.
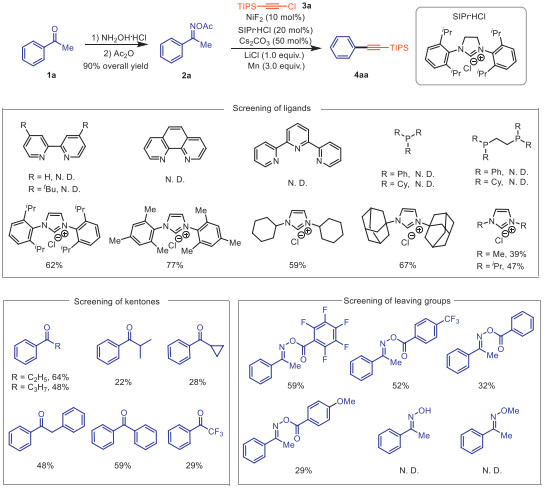
We start the investigations of cross-coupling of 1-phenylethan-1-one O-acetyl oxime 2a, which was derived from acetophenone 1a and hydroxylamine/acetic anhydride, with chloroethynyl-triisopropylsilane 3a (Table 1). After cautious and considerable optimization of all reaction parameters, we discovered that the composite use of NiF2, 1,3-bis-(2,6-diisopropylphenyl)imidazolinium chloride, Cs2CO3, LiCl and Mn in DMF gave triisopropyl(phenylethynyl)silane 4aa in 86% yield (entry 1). The use of CoCl2 and Fe(OTf)3 proved to be unsuccessful (entry 2), and other nickel catalysts, such as Ni(cod)2, were also effective for this transformation but with lower yields (entry 3). As anticipated, the influence of ligands is very important. Neither nitrogen ligands nor phosphine ligands can mediate the reaction effectively. N-Heterocyclic carbene (NHC) were found to be critical in the activation of C–C bonds. The SIPr ligand can also provide 80% yields (entry 4), and other NHC ligands also have moderate to good yields (Scheme 2, up) [89]. The replacement of Cs2CO3 with CsF or Na2CO3 led to decreased yields (entries 5 and 6). When LiCl as the additive was added to the reaction, the yield increased, while alternatives like MgCl2 or LiBr proved to be less effective (entries 7–9) [92–94]. In particularly, no reaction occurs when B2Pin2 is used as an organic reductant (entry 10). Then, the screenings of solvents were conducted, demonstrating that the reaction showed the best reactivity in DMF (entry 11). There was almost no product when the temperature was reduced to room temperature (entry 12), while high temperatures would lead to self-coupling products (entry 13). It should be noted that the metal catalyst, ligands and reducing agent were important in control experiments, as no reaction occurred in their absence (entry 14). Next, we screened the aryl ketone species and leaving groups because of their importance in this transformation. The steric and electronic effects on the alkyl side of acetophenone are evident. When propiophenone or butyrone was used as the substrate, the yield of the desired alkynylation product was 64% and 48%, respectively. Isobutyrophenone gave a yield of 22%, while cyclopropyl phenyl ketone afforded a yield of 28%. When 2-phenylacetophenone, benzophenone or 2,2,2-trifluoro-1-phenylethan-1-one was employed, the yield decreased to 48%, 59% and 29%. Finally, the screening for leaving groups of arylketone oxime esters suggested that most groups are compatible, but electron-rich groups perform better than electron-poor groups. The best choice among them is the acetoxy group, while the reaction fails to occur when using 1-phenylethan-1-one oxime or 1-phenylethan-1-one O-methyl oxime (Scheme 2, down).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||||||||||||||||||||||||||||||||||||||||
| Entry | Variation of optimal conditions | Yield of 4aa (%) | |||||||||||||||||||||||||||||||||||||||
| 1 | No | 86 | |||||||||||||||||||||||||||||||||||||||
| 2 | CoCl2 or Fe(OTf)3 instead of NiF2 | 0 | |||||||||||||||||||||||||||||||||||||||
| 3 | Ni(cod)2 instead of NiF2 | 71 | |||||||||||||||||||||||||||||||||||||||
| 4 | SIPr instead of SIPr.HCl and Cs2CO3 | 80 | |||||||||||||||||||||||||||||||||||||||
| 5 | CsF instead of Cs2CO3 | 35 | |||||||||||||||||||||||||||||||||||||||
| 6 | Na2CO3 instead of Cs2CO3 | 24 | |||||||||||||||||||||||||||||||||||||||
| 7 | without LiCl | 79 | |||||||||||||||||||||||||||||||||||||||
| 8 | MgCl2 instead of LiCl | 69 | |||||||||||||||||||||||||||||||||||||||
| 9 | LiBr instead of LiCl | 42 | |||||||||||||||||||||||||||||||||||||||
| 10 | B2Pin2 instead of Mn | 0 | |||||||||||||||||||||||||||||||||||||||
| 11 | DMSO as solvent | 66 | |||||||||||||||||||||||||||||||||||||||
| 12 | RT instead of 100 ℃ | < 1 | |||||||||||||||||||||||||||||||||||||||
| 13 | 120 ℃ instead of 100 ℃ | 48 | |||||||||||||||||||||||||||||||||||||||
| 14 | without catalyst, ligand or reductant | 0 | |||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: 2a (0.2 mmol), 3a (0.4 mmol), catalyst (10 mol%), ligand (20 mol%), base (50 mol%), additive (1.0 equiv.), reductant (3.0 equiv.) and solvent (1.0 mL), at 100 ℃ for 12 h under N2. GC yields. SIPr ·HCl = 1,3-Bis-(2,6-diisopropylphenyl)imidazolinium chloride, bpy = 2,2′-bipyridine, IPr·HCl = 1,3-diisopropylimidazolium chloride, DMF = N,N-dimethylformamide, DMSO = dimethyl sulfoxide, RT = room temperature. | |||||||||||||||||||||||||||||||||||||||||
Having optimized the reaction conditions, the substrates scope of this reductive alkynylation was then explored. Initially, various chloroalkynes were investigated (Scheme 3). Fortunately, good functional group compatibility was observed when alkyne starting materials were substituted with silicon, aryl or alkyl groups. tert-Butylsilyl (4ba), triethylsilyl (4bb), and trime-thylsilyl (4bc) substrates were converted to the corresponding functionalized products in high yields. The group compatibility of the aryl-substituted alkynyl chlorides unit was also examined, and a wide range of functional groups such as fluorine (4bd), naphthyl (4be) and heterocyclic (4bf-4bg) moieties were compatible with the reaction. Several functional groups can be appended onto the alkyl chain of the chloroalkynes, including N-phthalimide ester (4bh), cyanide (4bi) and butyl (4bj). Significantly, the reaction can also occur smoothly with the functional group containing free hydrogen, including -OH (4bk-4bm) and -NH2 (4bn). Furthermore, material molecules ferrocene (4bo), and biologically active alkynes, for instance, pargyline (4bp), ethisterone (4bq) and ethynyl estradiol (4br) were identified as viable substrates (Scheme 3A).
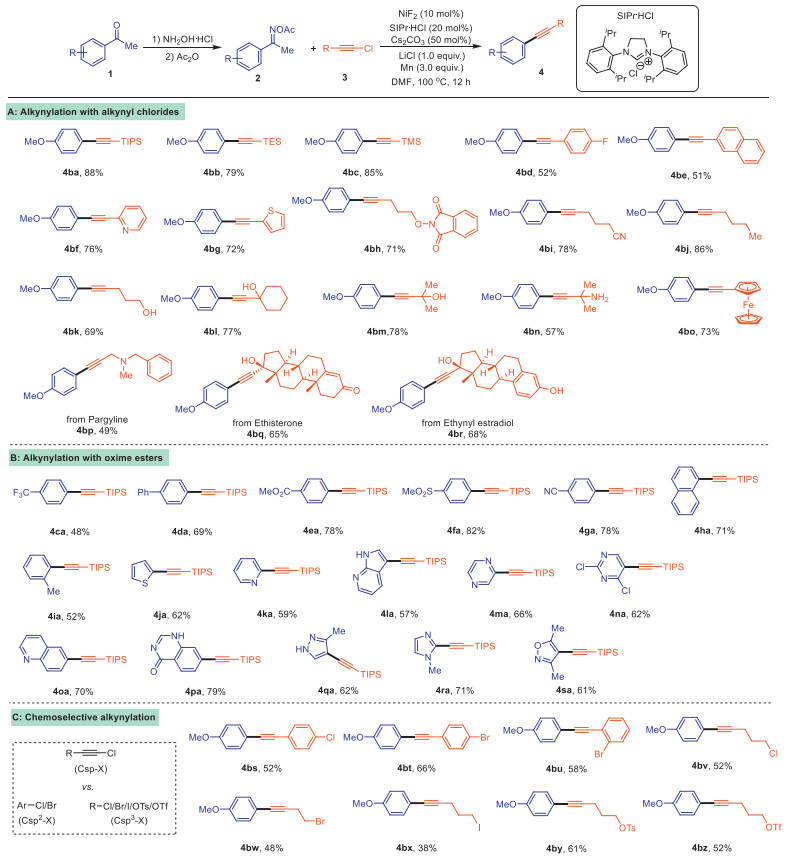
To explore the effect of substituents on ketoimines from ketones, we investigated a variety of acetophenone derivatives. As shown in Scheme 3, various aryl or heterocyclic acetophenones were converted to arylketone oxime esters, which can react well with (chloroethynyl) triisopropylsilane 3a. Generally, this method was also compatible with a wide range of substrates with electron-donating and -withdrawing functional groups (4ca-4ia). Notably, for those containing trifluoromethyl (4ca), ester (4ea), sulfonyl (4fa) and cyanide (4ga) were also well tolerated. Meanwhile, the high hindrance substrate 2′-methylacetophenone (4ia) can also be obtained with an 88% yield. Next, we focused on examination of heteroaryl substrates and found that the substrates with thiophene (4ja), pyridine (4ka), pyrrolidine (4la), pyrazine (4ma), pyrimidine (4na), quinoline (4oa), oxazoline (4pa), pyrazol (4qa), imidazole (4ra), isoxazole (4sa) were successful transformed to the desired products in moderate to good yields under the optimized conditions (Scheme 3B). Unfortunately, ketoimines obtained from alkyl-substituted ketones, such as heptan-2-one or cyclohexanone, failed to furnish the corresponding products under the current reaction conditions. And this reaction shown a poor selectivity when both sides of the ketone are aryl groups.
Satisfyingly, this reaction also exhibited a high chemo-selectivity of tolerance toward aryl chloride (4bs), aryl bromides bearing ortho or para substituents (4bt-4bu), alkyl chloride (4bv), alkyl bromide (4bw), alkyl iodide (4bx), alkyl tosylate (4by) and alkyl triflate (4bz), which are potential reaction sites for further transformation. The reactions were highly selectively occurred at the C(sp)–Cl position, with all other C(sp2)/C(sp3)–X groups untouched (Scheme 3C).
Noteworthily, it has been suggested that construct C(sp2)–C(sp) bond in biologically active complex molecules could change the pharmacokinetic properties and enhanced targeting of drugs, which can improve the clinical success of potential drug molecules. As such, to highlight the synthetic utility of reductive alkynylation transformation, we selected many complex molecules as the substrates for late-stage diversification. Fortunately, good yields and high selectivity were observed. Biologically active molecules including antidiabetic Canagliflozin(7aa) and Empagliflozin (7ba), antitumor Lapatinib (7ca), anticancer Erlotinib (7da) and Trametinib (7ea and 7fa), antidote Idoxuridine (7ga) and Furosemide (7ha), and C-Glycoside derivatives (7ia and 7ja) were recognized as worthwhile substrates. Moreover, the complexes from uricosuric Febuxostat (7ka), anticancer Bexarotene (7la), Vitamin Adapalene (7ma) and Retinoic acid (7na), Inhibitor Ciprofibrate (7oa) antidiabetic Repaglinid (7pa), and nature product peptide (7qa) also efficaciously construct the targeted C(sp2)–C(sp) bond, which illustrates the capability to organize the late-stage alkynylation of complex structures (Scheme 4).

Several experiments were carried out to get insight into the conceivable mechanism of this conversion. In order to clarify whether the reaction was carried out through radical chain or sequential reduction, radical quenchers 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) and butylated hydroxytoluene (BHT) were added to the reaction system. However, a significant amount of product was still produced (for details see Supporting information), suggesting that the radical intermediates might not be involved in the reaction. When a milder reductant Zn was used, the reaction gave a decreased yield of the desired alkynylation products 4ha, while the direct C–N coupling byproduct ynimine 8 was obtained in 12% yield, and the using of Fe resulted in trace amount of products, demonstrating that the choice of metal reductant with appropriate reduction potential is important for this transformation (Scheme 5A). Next, the preparation of alkynyl organonickel complex 9 was conducted, and the decarbonylative product 4ha could be obtained in 41% yield when nickel complex 9 was used in the reaction [95]. This finding indicated that the nickel complex 9 might a reaction intermediate in the C–C activation process. Furthermore, when the competitive experiment with nickel complex 9, 10 and 2h in each 1.0 equiv. was performed, 30% of 4ha and 21% of 12 products were obtained, indicating that the nickel intermediates 9 could act as both pivotal material and catalyst (Scheme 5C). The stoichiometric reaction with 1.0 equiv. of Ni(cod)2 and 2.0 equiv. of ligand generated 66% of the desired product 4ha in the presence of Mn, while only a 30% yield of 4ha was observed without Mn reductant (Scheme 5D), suggesting that the Mn reductive process is probably involved in the catalytic cycle. Additionally, the possible path involving the initial 1,2-shift of the aryl group was rejected because the supposed intermediate, acetanilide 12 was ineffective under the standard conditions [96]. Meanwhile, as a direct C–N cross-coupling product, ynimine 8 is also excluded (Scheme 5E) [97,98]. The experimental profiles of these two intermediates further demonstrated the existence of β-carbon elimination processes during the reaction process. Finally, the range of alkynes was examined. As another alkyl electrophilic, bromoalkyne showed a moderate yield. However, the reaction could not proceed for diphenylacetylene and phenylacetylene. Therefore, we speculate that this reductive alkynylation strategy does not involve addition/elimination or Socoupling via transmetalation process (Scheme 5F).
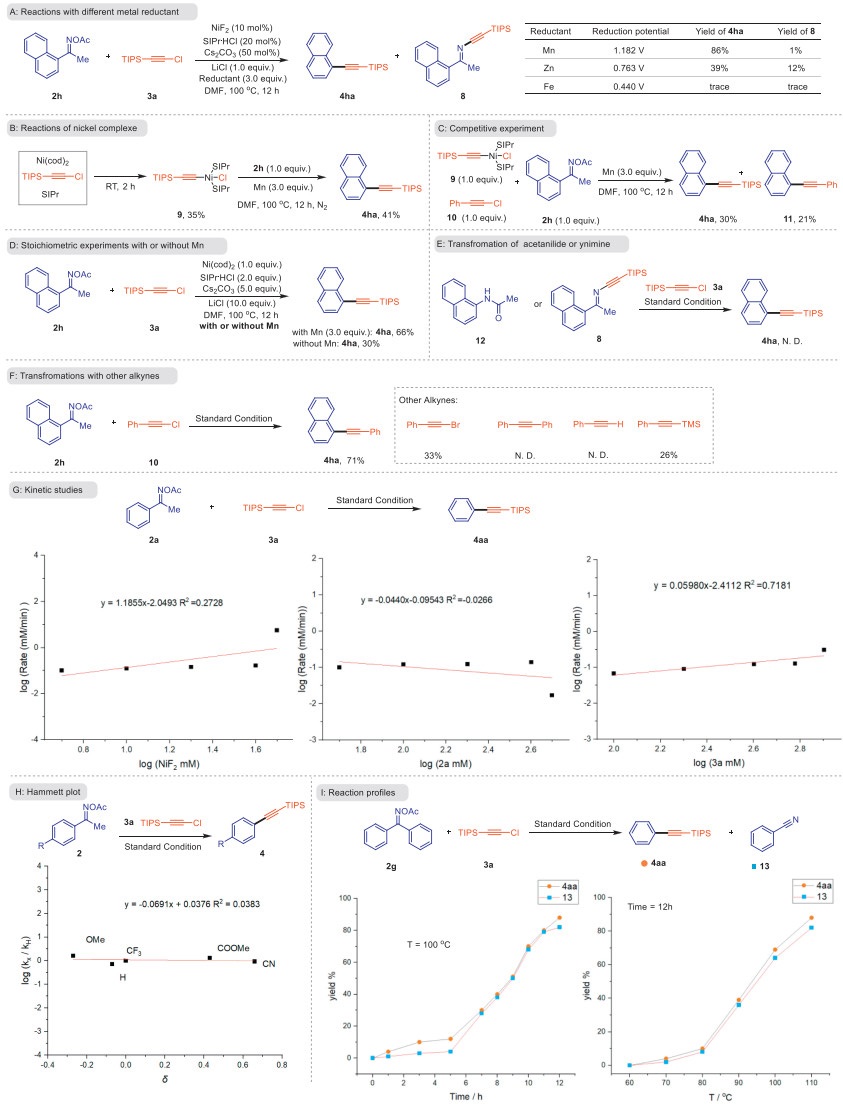
Then we organized kinetic researches to explore possible influencing factors (Schemes 5G-I). A first-order confidence with showing respect for the consistence of metal catalyst, and zero-order for ketoimines and chloroalkynes were discovered, signposting that an intermediate accumulated from both reagents might be involved in the turnover-limiting step (Scheme 5G) [99]. Next, a ρ value approaching zero was attained from Hammett's studies, suggesting the charge distribution on the substrate does not have a strong effect on the rate of the reaction, which is reliable with the results of kinetic research (Scheme 5H) [100]. Finally, the effects of time and temperature on the alkynylation products and benzonitrile byproducts 13 obtained through elimination were studied. Overall, the concentration of these two substances are similar as the reaction progressed, further confirming the existence and importance of β-carbon eliminating in the reaction (Scheme 5I).
DFT calculations were further performed to investigate the mechanism for this nickel-catalyzed reductive alkynylation of oximes. The computed energy profiles for the reaction of alkyne 3c with oxime 2b are shown in Scheme 6. Due to the back-donation effect [101,102], the complex of Ni(0) with chloroalkyne (INT1, Scheme 6A) was identified as the most stable species among a series of possible catalyst-substrate complexes (see details in Fig. S13 in Supporting information). The C(alkynyl)–Cl oxidative addition requires a barrier of 24.9 kcal/mol (TS1), which is 5.0 kcal/mol lower than that of N–O oxidative cleavage (TS2), indicating the reaction is initiated by chloroalkynes rather than oximes. Compared with the C(sp)–Cl oxidative addition, the cleavages of C(sp3)–Cl and C(sp2)–Cl are much more difficult (Fig. S14 in Supporting information), which is in line with the excellent chemo-selectivity of this reaction (Scheme 3C).
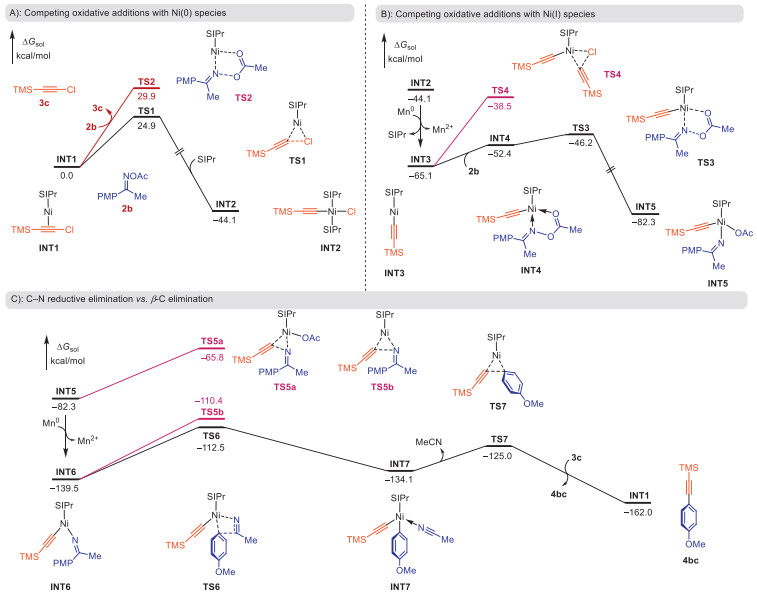
In the presence of a reductant, the formed alkynyl-Ni(Ⅱ) intermediate (INT2) can be reduced to alkynyl-Ni(Ⅰ) species (INT3) (Scheme 6B, see details in Fig. S15 in Supporting information). Subsequently, both chloroalkyne and oxime could react with INT3 via C–Cl and N–O oxidative additions, respectively. The computation shows that the N–O cleavage (TS3) is more favorable than the C–Cl cleavage (TS4), leading to the alkynyl-Ni(Ⅲ) intermediate (INT5). Based on INT5, the direct C–N reductive elimination via TS5a (ΔG‡ = 16.5 kcal/mol, Scheme 6C) affords the undesired ynimine product. However, this process can be suppressed by reductant. The reduction of alkynyl-Ni(Ⅲ) (INT5) to alkynyl-Ni(Ⅱ) (INT6) is thermodynamically favored (Fig. S15). This is consistent with the control experiment in which the yield of ynimine increases when employing a weaker reductant (Scheme 5A). Based on INT6, the C(aryl)–C(imine) bond is cleaved via β-carbon elimination (TS6), requiring a barrier of 27.0 kcal/mol. This process is superior to the C–N reductive elimination (TS5b, ΔG‡ = 29.1 kcal/mol). The ensuing C(alkynyl)–C(aryl) reductive elimination (TS7) delivers the desired alkynylation product (4bc).
Taken together, the β-carbon elimination represents the rate-determining step in this reaction. The overall catalytic cycle involves Ni(0)/Ni(Ⅰ)/Ni(Ⅱ)/Ni(Ⅲ) species (the pathway shown in black in Scheme 6). The Mn reductant plays a crucial role in modulating the transformations among these Ni species, facilitating the formation of key alkynyl-Ni(Ⅰ) (INT3) and alkynyl-Ni(Ⅱ) (INT6) intermediates. This renders the process of alkynylation of aryl ketones superior to a series of competing unproductive pathways.
In conclusion, we have developed a nickel-catalyzed approach for reductive alkynylation of ketoimines from aryl ketones with chloroalkynes via the cleavage of unstrained C–C bond. This strategy shows excellent compatibility with sensitive halides, free hydroxyl and heteroaryl groups. In this method, β-carbon elimination through the nickel/NHC system is a pivotal elementary progress in the mechanism, providing a new platform for the cross electrophile coupling reaction. The late-stage alkynylation of complex molecules also demonstrated the potential utility of this protocol. Kinetic studies and theoretical calculations indicated that the β-carbon elimination was involved as the rate-determining step. Studies aimed at expanding on the cleavage of C–C bond for cross electrophile coupling will be continuously explored in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Tian-Zhang Wang: Writing – original draft, Methodology, Investigation. Le-Yu Tang: Methodology. Yu-Qiu Guan: Investigation. Lingfei Hu: Investigation. Gang Lu: Project administration. Yu-Feng Liang: Project administration.
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Nos. 22001147, 22371171), Taishan Scholars Project of Shandong Province (No. tsqn202103027), Distinguished Young Scholars of Shandong Province (Overseas) (No. 2022HWYQ-001), and Qilu Youth Scholar Funding of Shandong University.
Supplementary material associated with this article can be found, in the online version, at doi:
F. Chen, T. Wang, N. Jiao, Chem. Rev. 114 (2014) 8613–8661. doi: 10.1021/cr400628s
F. Song, B. Wang, Z.J. Shi, Acc. Chem. Res. 56 (2023) 2867–2886. doi: 10.1021/acs.accounts.3c00230
J. Xie, H. Jin, A.S.K. Hashmi, Chem. Soc. Rev. 46 (2017) 5193–5203. doi: 10.1039/C7CS00339K
H. Yang, Z. Zhou, C. Tang, F. Chen, Chin. Chem. Lett. 35 (2024) 109257. doi: 10.1016/j.cclet.2023.109257
B. Eftekhari-Sis, M. Zirak, Chem. Rev. 115 (2015) 151–264. doi: 10.1021/cr5004216
F. Song, B. Wang, Z. J Shi, Acc. Chem. Res. 56 (2023) 2867–2886. doi: 10.1021/acs.accounts.3c00230
M. Rubin, M. Rubina, V. Gevorgyan, Chem. Rev. 107 (2007) 3117–3179. doi: 10.1021/cr050988l
B. Yuan, D. Ding, C. Wang, ACS Catal. 12 (2022) 4261–4267. doi: 10.1021/acscatal.2c00677
R. Vicente, Chem. Rev. 121 (2021) 162–226. doi: 10.1021/acs.chemrev.0c00151
Y. Xue, G. Dong, Acc. Chem. Res. 55 (2022) 2341–2354. doi: 10.1021/acs.accounts.2c00400
D.S. Kim, W.J. Park, C.H. Jun, Chem. Rev. 117 (2017) 8977–9015. doi: 10.1021/acs.chemrev.6b00554
Y.F. Liang, N. Jiao, Acc. Chem. Res. 50 (2017) 1640–1653. doi: 10.1021/acs.accounts.7b00108
H. Wang, I. Choi, T. Rogge, N. Kaplaneris, L. Ackermann, Nat. Catal. 1 (2018) 993–1001. doi: 10.1038/s41929-018-0187-1
X. Cheng, A. Lei, T.S. Mei, et al., CCS Chem. 4 (2022) 1120–1152. doi: 10.31635/ccschem.021.202101451
S.H. Shi, Y. Liang, N. Jiao, Chem. Rev. 121 (2021) 485–505. doi: 10.1021/acs.chemrev.0c00335
H. Lu, T.Y. Yu, P.F. Xu, H. Wei, Chem. Rev. 121 (2021) 365–411. doi: 10.1021/acs.chemrev.0c00153
Y.F. Liang, M. Bilal, L.Y. Tang, et al., Chem. Rev. 123 (2023) 12313–12370. doi: 10.1021/acs.chemrev.3c00219
W. Shang, R. Shi, D. Niu, Chin. J. Chem. 41 (2023) 2217–2236. doi: 10.1002/cjoc.202300153
N.R. Candeias, R. Paterna, P.M.P. Gois, Chem. Rev. 116 (2016) 2937–2981. doi: 10.1021/acs.chemrev.5b00381
J. Zhou, G. Xu, Y. Ni, ACS Catal. 10 (2020) 10954–10966. doi: 10.1021/acscatal.0c02646
Y. Xu, X. Qi, P. Zheng, et al., Nature 567 (2019) 373–378. doi: 10.1038/s41586-019-0926-8
J. Cao, H. Wu, Q. Wang, J. Zhu, Nat. Chem. 13 (2021) 671–676. doi: 10.1038/s41557-021-00698-y
F. Song, T. Gou, B.Q. Wang, Z.J. Shi, Chem. Soc. Rev. 47 (2018) 7078–7115. doi: 10.1039/c8cs00253c
Z.Q. Lei, H. Li, Y. Li, et al., Angew. Chem. Int. Ed. 51 (2012) 2690–2694. doi: 10.1002/anie.201107136
Z.Q. Lei, F. Pan, H. Li, et al., J. Am. Chem. Soc. 137 (2015) 5012–5020. doi: 10.1021/ja512003d
A. Dermenci, R.E. Whittaker, G. Dong, Org. Lett. 15 (2013) 2242–2245. doi: 10.1021/ol400815y
A. Dermenci, R.E. Whittaker, Y. Gao, et al., Chem. Sci. 6 (2015) 3201–3210. doi: 10.1039/C5SC00584A
R. Somerville, R. Martin, Angew. Chem. Int. Ed. 56 (2017) 6708–6710. doi: 10.1002/anie.201702188
T. Morioka, A. Nishizawa, T. Furukawa, M. Tobisu, N. Chatani, J. Am. Chem. Soc. 139 (2017) 1416–1419. doi: 10.1021/jacs.6b12293
T.T. Zhao, W.H. Xu, Z.J. Zheng, P.F. Xu, H. Wei, J. Am. Chem. Soc. 140 (2018) 586–589. doi: 10.1021/jacs.7b11591
J.H. Guo, Y. Liu, X.C. Lin, et al., Angew. Chem. Int. Ed. 60 (2021) 19079–19084. doi: 10.1002/anie.202106709
M.E. O'Reilly, S. Dutta, A.S. Veige, Chem. Rev. 116 (2016) 8105–8145. doi: 10.1021/acs.chemrev.6b00054
M.D.R. Lutz, D. Morandi, Chem. Rev. 121 (2021) 300–326. doi: 10.1021/acs.chemrev.0c00154
A.A. Tabolin, S.L. Ioffe, Chem. Rev. 114 (2014) 5426–5476. doi: 10.1021/cr400196x
H. Huang, X. Ji, W. Wu, H. Jiang, Chem. Soc. Rev. 44 (2015) 1155–1171. doi: 10.1039/C4CS00288A
D.D. Dong, J.C. Song, S.H. Yang, et al., Chin. Chem. Lett. 33 (2022) 1199–1206. doi: 10.1016/j.cclet.2021.08.067
B. Zhao, Z. Shi, Angew. Chem. Int. Ed. 56 (2017) 12727–12731. doi: 10.1002/anie.201707181
D. Ding, C. Wang, ACS Catal. 8 (2018) 11324–11329. doi: 10.1021/acscatal.8b03930
W. Ai, Y. Liu, Q. Wang, Z. Lu, Q. Liu, Org. Lett. 20 (2018) 409–412. doi: 10.1021/acs.orglett.7b03707
S. Cui, X. Wu, W. Ma, et al., Green Syn. Cataly. 2 (2021) 307–310.
Z. Li, R.O. Torres-Ochoa, Q. Wang, J. Zhu, Nat. Commun. 11 (2020) 403–409. doi: 10.1038/s41467-020-14292-2
P.Z. Wang, Y. Gao, J. Chen, et al., Nat. Commun. 12 (2021) 1815–1825. doi: 10.1038/s41467-021-22127-x
C.H. Jun, H. Lee, S.G. Lim, J. Am. Chem. Soc. 123 (2001) 751–752. doi: 10.1021/ja0033537
Y. Xia, G. Lu, P. Liu, G. Dong, Nature 539 (2016) 546–550. doi: 10.1038/nature19849
R. Zhang, T. Yu, G. Dong, Science 382 (2023) 951–957. doi: 10.1126/science.adk1001
H. Li, B. Ma, Q.S. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 14388–14393. doi: 10.1002/anie.202006740
H. Li, M.L. Wang, Y.W. Liu, et al., ACS Catal. 12 (2022) 82–88. doi: 10.1021/acscatal.1c04010
Z.Y. Wang, X. Zhang, W.Q. Chen, et al., Angew. Chem. Int. Ed. 63 (2024) e202319773. doi: 10.1002/anie.202319773
D.J. Weix, Acc. Chem. Res. 48 (2015) 1767–1775. doi: 10.1021/acs.accounts.5b00057
J. Liu, Y. Ye, J.L. Sessler, H. Gong, Acc. Chem. Res. 53 (2020) 1833–1845. doi: 10.1021/acs.accounts.0c00291
X. Pang, P.F. Su, X.Z. Shu, Acc. Chem. Res. 55 (2022) 2491–2509. doi: 10.1021/acs.accounts.2c00381
Q. Pan, Y. Ping, W. Kong, Acc. Chem. Res. 56 (2023) 515–535. doi: 10.1021/acs.accounts.2c00771
G. Li, T. Wang, F. Fei, et al., Angew. Chem. Int. Ed. 55 (2016) 3491–3495. doi: 10.1002/anie.201511321
Z. Li, K.F. Wang, X. Zhao, et al., Nat. Commun. 11 (2020) 5036–5047. doi: 10.1038/s41467-020-18834-6
Y. Dai, F. Wang, S. Zhu, L. Chu, Chin. Chem. Lett. 33 (2022) 4074–4078. doi: 10.1016/j.cclet.2021.12.050
Q.Q. Pan, L. Qi, X. Pang, X.Z. Shu, Angew. Chem. Int. Ed. 62 (2023) e202215703. doi: 10.1002/anie.202215703
L. Xi, L. Du, Z. Shi, Chin. Chem. Lett. 33 (2022) 4287–4292. doi: 10.1016/j.cclet.2022.01.077
Y.Z. Li, N. Rao, L. An, et al., Nat. Commun. 13 (2022) 5539. doi: 10.1038/s41467-022-33159-2
Q. Lin, G. Ma, H. Gong, ACS Catal. 11 (2021) 14102–14109. doi: 10.1021/acscatal.1c04239
L. Qi, X. Pang, K. Yin, et al., Chin. Chem. Lett. 33 (2022) 5061–5064. doi: 10.1016/j.cclet.2022.03.070
Y. Li, Y. Li, H. Shi, et al., Science 376 (2022) 749–753. doi: 10.1126/science.abn9124
Y. Gong, L. Su, Z. Zhu, Y. Ye, H. Gong, Angew. Chem. Int. Ed. 61 (2022) e202201662. doi: 10.1002/anie.202201662
X. Wu, A. Turlik, B. Luan, et al., Angew. Chem. Int. Ed. 61 (2022) e202207536. doi: 10.1002/anie.202207536
J. Lu, Y. Yao, L. Li, N. Fu, J. Am. Chem. Soc. 145 (2023) 26774–26782. doi: 10.1021/jacs.3c08839
W. Du, Q. Luo, Z. Wei, et al., Sci. China Chem. 66 (2023) 2785–2790. doi: 10.1007/s11426-023-1791-3
Y. Wang, Y. Ping, W. Kong, Chin. Chem. Lett. 34 (2023) 108453. doi: 10.1016/j.cclet.2023.108453
Q.Q. Pan, L. Qi, X. Pang, X.Z. Shu, Angew. Chem. Int. Ed. 62 (2023) e202215703. doi: 10.1002/anie.202215703
X. Ying, Y. Li, L. Li, C. Li, Angew. Chem. Int. Ed. 62 (2023) e202304177. doi: 10.1002/anie.202304177
J. Yang, Z. Gui, Y. He, S. Zhu, Angew. Chem. Int. Ed. 62 (2023) e202304713. doi: 10.1002/anie.202304713
L. Cheng, J. Liu, Y. Chen, H. Gong, Nat. Synth. 2 (2023) 364–372. doi: 10.1038/s44160-023-00239-0
Y.C. Luo, M.K. Wang, L.C. Yu, X. Zhang, Angew. Chem. Int. Ed. 62 (2023) e202308690. doi: 10.1002/anie.202308690
L. Wan, Y. Tong, X. Lu, Y. Fu, Chin. Chem. Lett. 35 (2024) 109283. doi: 10.1016/j.cclet.2023.109283
Q. Pan, K. Wang, W. Xu, et al., J. Am. Chem. Soc. 146 (2024) 15453–15463. doi: 10.1021/jacs.4c03745
Y.P. Shao, Z.M. Chi, Y.M. Liang, Sci. China Chem. 67 (2024) 1935–1940. doi: 10.1007/s11426-024-1949-1
P. Li, Z. Zhu, C. Guo, et al., Nat. Catal. 7 (2024) 412–421. doi: 10.1038/s41929-024-01118-3
S.C. Wang, L. Liu, M. Duan, et al., J. Am. Chem. Soc. 146 (2024) 30626–30636. doi: 10.1021/jacs.4c12324
D. Zeng, Z. Liu, G. Huang, Y. Wang, S. Zhu, Nat. Commun. 15 (2024) 7645. doi: 10.1038/s41467-024-52054-6
D. Sun, Y. Gong, Y. Wu, Y. Chen, H. Gong, Adv. Sci. 11 (2024) 2404301. doi: 10.1002/advs.202404301
F.F. Tong, Y.C. Luo, H.Y. Zhao, X.P. Fu, X. Zhang, Angew. Chem. Int. Ed. 63 (2024) e202417858.
J. Zhou, Y. He, Z. Liu, Y. Wang, S. Zhu, Adv. Sci. 11 (2024) 2306447. doi: 10.1002/advs.202306447
X.W. Chen, C. Li, Y.Y. Gui, et al., Angew. Chem. Int. Ed. 63 (2024) e202403401. doi: 10.1002/anie.202403401
S. Lu, Z. Hu, D. Wang, T. XU, Angew. Chem. Int. Ed. 63 (2024) e202406064. doi: 10.1002/anie.202406064
L. Ding, M. Wang, Y. Liu, et al., Angew. Chem. Int. Ed. 63 (2024) e202413557.
G.Y. Han, P.F. Su, Q.Q. Pan, X.Y. Liu, X.Z. Shu, Nat. Catal. 7 (2024) 12–20.
T.Z. Wang, Y.Q. Guan, T.Y. Zhang, Y.F. Liang. Adv. Sci. 11 (2024) 2306923. doi: 10.1002/advs.202306923
J.W. Wang, Q.W. Zhu, D. Liu, et al., Angew. Chem. Int. Ed. 63 (2024) e202413074. doi: 10.1002/anie.202413074
X.B. Liu, R.M. Liu, X.D. Bao, H.J. Xu, Y.F. Liang, Chin. Chem. Lett. 35 (2024) 109783. doi: 10.1016/j.cclet.2024.109783
X. Zhang, W. Su, H. Guo, et al., Angew. Chem. Int. Ed. 63 (2024) e202318613. doi: 10.1002/anie.202318613
X. Luo, W. Mao, C.F. Liu, et al., Nat. Synth. 3 (2024) 633–642. doi: 10.1038/s44160-024-00492-x
S. Xu, Y. Ping, M. Xu, et al., Nat. Chem. 16(2024) 2054–2065. doi: 10.1038/s41557-024-01629-3
C. Yoo, S. Bhattacharya, X. Yi, et al., Science 382 (2023) 815–820. doi: 10.1126/science.ade3179
X. Wang, G. Ma, Y. Peng, et al., J. Am. Chem. Soc. 140 (2018) 14490–14497. doi: 10.1021/jacs.8b09473
L. Huang, L.K.G. Ackerman, K. Kang, A.M. Parsons, D.J. Weix, J. Am. Chem. Soc. 141 (2019) 10978–10983. doi: 10.1021/jacs.9b05461
L.E. Ehehalt, O.M. Beleh, I.C. Priest, et al., Chem. Rev. 124 (2024) 13397–13569. doi: 10.1021/acs.chemrev.4c00524
S.H. Newman-Stonebraker, T.J. Raab, H. Roshandel, A.G. Doyle, J. Am. Chem. Soc. 145 (2023) 19368–19377. doi: 10.1021/jacs.3c06233
X. Mo, T.D.R. Morgan, H.T. Ang, D.G. Hall, J. Am. Chem. Soc. 140 (2018) 5264–5271. doi: 10.1021/jacs.8b01618
R. Lavernhe, R.O. Torres-Ochoa, Q. Wang, J. Zhu, Angew. Chem. Int. Ed. 60 (2021) 24028–24033. doi: 10.1002/anie.202110901
Y.M. Cai, X.T. Liu, L.L. Xu, M. Shang, Angew. Chem. Int. Ed. 63 (2024) e202315222. doi: 10.1002/anie.202315222
C.X. Liu, P.P. Xie, F. Zhao, et al., J. Am. Chem. Soc. 145 (2023) 4765–4773. doi: 10.1021/jacs.2c13542
B.E. Nadeau, D.D. Beattie, E.K.J. Lui, et al., Organometallics 42 (2023) 2326–2334. doi: 10.1021/acs.organomet.3c00199
X. Ren, Y. Lu, G. Lu, Z.X. Wang, Org. Lett. 22 (2020) 2454–2459. doi: 10.1021/acs.orglett.0c00674
Y.Q. Zhang, L. Hu, L. Yuwen, G. Lu, Q.W. Zhang, Nat. Catal. 6 (2023) 487–494. doi: 10.1038/s41929-023-00966-9

Scheme 1 Transition metal-catalyzed unstrained C–C bond activation for cross coupling.

Scheme 3 Scope of reductive alkynylation reaction of chloroalkynes. Reaction conditions: 2 (0.2 mmol), 3 (0.4 mmol), NiF2 (10 mol%), SIPr·HCl (20 mol%), Cs2CO3 (50 mol%), LiCl (1.0 equiv.), Mn (3.0 equiv.) and DMF (1 mL), at 100 ℃ for 12 h under N2. Isolated yields.

Scheme 4 Late-stage reductive alkynylation of complex molecules. Reaction conditions: 6 (0.2 mmol), 2a (0.4 mmol), NiF2 (10 mol%), SIPr·HCl (20 mol%), Cs2CO3 (50 mol%), LiCl (1.0 equiv.), Mn (3.0 equiv.) and DMF (1 mL), at 100 ℃ for 12 h under N2. Isolated yields.
Table 1. Establishing the reductive alkynylation reaction.a
|
|||||||||||||||||||||||||||||||||||||||||
| Entry | Variation of optimal conditions | Yield of 4aa (%) | |||||||||||||||||||||||||||||||||||||||
| 1 | No | 86 | |||||||||||||||||||||||||||||||||||||||
| 2 | CoCl2 or Fe(OTf)3 instead of NiF2 | 0 | |||||||||||||||||||||||||||||||||||||||
| 3 | Ni(cod)2 instead of NiF2 | 71 | |||||||||||||||||||||||||||||||||||||||
| 4 | SIPr instead of SIPr.HCl and Cs2CO3 | 80 | |||||||||||||||||||||||||||||||||||||||
| 5 | CsF instead of Cs2CO3 | 35 | |||||||||||||||||||||||||||||||||||||||
| 6 | Na2CO3 instead of Cs2CO3 | 24 | |||||||||||||||||||||||||||||||||||||||
| 7 | without LiCl | 79 | |||||||||||||||||||||||||||||||||||||||
| 8 | MgCl2 instead of LiCl | 69 | |||||||||||||||||||||||||||||||||||||||
| 9 | LiBr instead of LiCl | 42 | |||||||||||||||||||||||||||||||||||||||
| 10 | B2Pin2 instead of Mn | 0 | |||||||||||||||||||||||||||||||||||||||
| 11 | DMSO as solvent | 66 | |||||||||||||||||||||||||||||||||||||||
| 12 | RT instead of 100 ℃ | < 1 | |||||||||||||||||||||||||||||||||||||||
| 13 | 120 ℃ instead of 100 ℃ | 48 | |||||||||||||||||||||||||||||||||||||||
| 14 | without catalyst, ligand or reductant | 0 | |||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: 2a (0.2 mmol), 3a (0.4 mmol), catalyst (10 mol%), ligand (20 mol%), base (50 mol%), additive (1.0 equiv.), reductant (3.0 equiv.) and solvent (1.0 mL), at 100 ℃ for 12 h under N2. GC yields. SIPr ·HCl = 1,3-Bis-(2,6-diisopropylphenyl)imidazolinium chloride, bpy = 2,2′-bipyridine, IPr·HCl = 1,3-diisopropylimidazolium chloride, DMF = N,N-dimethylformamide, DMSO = dimethyl sulfoxide, RT = room temperature. | |||||||||||||||||||||||||||||||||||||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: