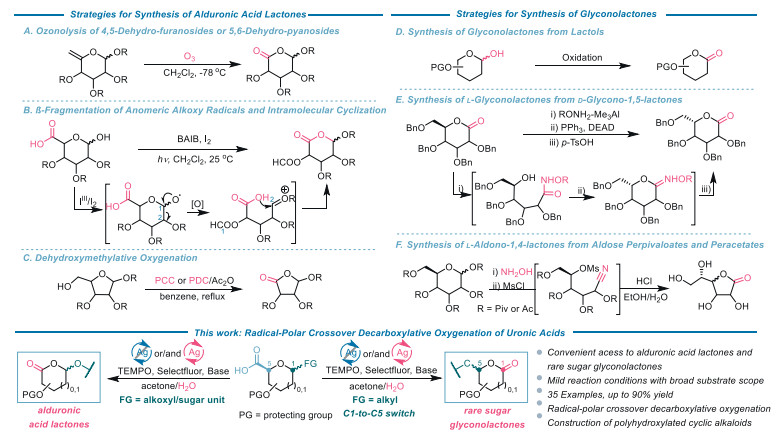
Scheme 1.
Strategies for the synthesis of alduronic acid lactones and glyconolactones.
Synthesis of alduronic acid lactones and rare sugar glyconolactones via decarboxylative oxygenation of uronic acids: Construction of polyhydroxylated fused-ring alkaloids
Pengwei Chen , Xian Ma , Ni Song , Jianjun Wang , Han Ding , Peng Wang , Hongzhi Cao , Xue-Wei Liu , Zhihua Lv , Ming Li

Alduronic acid lactones are a mesmerizing and versatile class of synthons because of the presence of an ester linkage, an acetal moiety, and multiple stereochemically defined contiguous hydroxy groups that are applicable to diverse functional group transformations [1]. Such constructs have been extensively employed in the synthesis of iminosugars [2], carbocyclic nucleosides [3], and carbasugar derivatives [4] of biological importance. Current strategies for preparing alduronic acid lactones could be divided into three classes (Schemes 1A–C). The first method involves ozonolysis of either 4,5-dehydro-furanosides or 5,6-dehydro-pyranosides, which are usually prepared by base-mediated elimination of sugar-based primary halides or tosylates (Scheme 1A) [5]. This procedure is the most popular protocol for making alduronic acid lactones so far. The second elegant approach to alduronic acid lactones was introduced by the Suárez and co-workers. This approach entails a [bis(acetoxy)iodo]benzene (BAIB)/iodine-promoted tandem reaction. The transformation proceeds through β-fragmentation of the anomeric alkoxy radicals followed by the generation of a oxocarbenium ion, and subsequent intramolecular cyclization (Scheme 1B) [6]. The third method capitalized on stoichiometric chromium salt-mediated dehydroxymethylative oxygenation which shortened the carbon chain by one carbon in refluxing benzene (Scheme 1C) [7]. This reaction offers an alternative disconnection strategy to construct lactone rings and has been adopted to synthesize natural products embedded by γ- or δ-lactone moiety [8,9].
Similar to alduronic acid lactones, glyconolactones are also useful chiral sources in organic synthesis [10]. The oxidation of lactols is the most commonly used method to prepare glyconolactones (Scheme 1D) [10,11]. However, this strategy precludes the synthesis of rare D- and L-sugar glyconolactones because of the scarcity of starting materials. Intense efforts have been devoted to the conversion of easily available D-sugar derivatives into uncommon D- and L-glyconolactones. The Ikegami group [12] reported an innovative method for converting D-glycono-1,5-lactones into L-counterparts (Scheme 1E). The transformation proceeded through MeAl3-mediated alkoxyamination of D-lactones and intramolecular Mitsunobu reaction of the resultant δ-hydroxyalkoxamates. This process furnished L-glyconolactone-based oxime, which upon hydrolysis afford L-lactones. Unprotected L-aldono-1,4-lactones have been accomplished from D-aldose perpivaloates and peracetates (Scheme 1F) [13]. The synthesis involved preparation of the key 5-O-methylsulfonyl-d-glyconitrile intermediate, neighboring group-assisted inversion of the C-5 configuration, acid-promoted hydrolysis of the cyano and ester groups, and spontaneous cyclization through the ester bond formation. Additionally, gluconolactone has been synthesized through enzymatic method [14].
Despite these remarkable advances, efficient and broadly applicable approaches to alduronic acid lactones and rare sugar glyconolactones remain highly desirable to meet the growing demand for such structural motifs. In continuation of our interest in radical dehydroxymethylative fluorination [15,16] and decarboxylative fluorination of sugars [17,18], we here report decarboxylative oxygenation reaction of readily accessible uronic acids. The reaction enables convenient access to structurally diverse and synthetically important alduronic acid lactones that are decorated with various protecting groups and substituents. The method also allows the synthesis of various rare D- and L-glyconolactones with significant synthetic potential using readily available α- and β-d-C-alkyl-glycuronic acids as the starting materials. These transformations proceed well under mild reaction conditions by the combined action of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and Selectfluor in the absence/presence of catalytic amounts of Ag2CO3. The 18O-labeling experiments and the identification of the key intermediates suggest that a radical-polar crossover process operates in the reaction. To further showcase the potential of the developed method, syntheses of the polyhydroxylated cyclic alkaloids, which possess indolo[2,3-a]- or benzo[a]quinolizidine fused-ring system or castanospermine-type framework, have been achieved. These syntheses used D-xyluronic acid lactone, arylethylamines, and 5-(trimethylsilyl)pent‑3-en-1-amine as the building blocks, and proceeded with aminolysis of the lactone and cyclization induced by the N-acyliminium ion as the key steps.
Discovery of decarboxylative oxygenation of uronic acids. Trapping experiments utilizing TEMPO as a radical scavenger are widely recognized as a valuable tool for validating the radical mechanism involved in a reaction [19]. During our mechanistic studies on silver-mediated oxidative decarboxylative fluorination of uronic acids [17], we initially attempted to synthesize adduct 2 as an evidence supporting the proposed radical pathway (Table 1). The experiment involved the addition of 1.0 equiv. of TEMPO to a reaction mixture designed for decarboxylative fluorination, consisting of 1.0 equiv. of glucuronic acid 1a, 5.0 equiv. of Selectfluor, 0.5 equiv. of Ag2CO3, and 5.0 equiv. of KF·2H2O in aqueous acetone. Unexpectedly, the reaction resulted in the formation of xyluronic acid lactone 3a in 52% yield, accompanied by the formation of hemiacetal 3a' in 13% yield while the expected 2 was not detected (Table 1, entry 1). This serendipitous discovery sparked our interest in synthesizing aldouronic acid lactones through decarboxylative oxygenation of uronic acids because this transformation offers a novel pathway to access highly valuable lactones and broadens the potential of uronic acids in organic synthesis.
 DownLoad:
CSV
DownLoad:
CSV
 |
|||||
| Entry | Ag2CO3 (equiv.) | Base (equiv.) | Organic solvent | Yield (%)b | |
| 3a | 3a' | ||||
| 1 | 0.5 | KF·2H2O (5.0) | Acetone | 52 | 13 |
| 2 | 0.5 | — | Acetone | NR | — |
| 3 | — | KF·2H2O (5.0) | Acetone | 41 | 11 |
| 4 | — | NaHCO3 (5.0) | Acetone | 65 | 14 |
| 5 | — | NaHCO3 (5.0) | MeCN | 47 | 16 |
| 6 | — | NaHCO3 (5.0) | DMF | 7 | ND |
| 7 | — | NaHCO3 (5.0) | DMSO | NR | — |
| 8 | — | CsHCO3 (5.0) | Acetone | 80 | ND |
| 9 | — | KHCO3 (5.0) | Acetone | 83 | ND |
| 10 | — | KHCO3 (8.0) | Acetone | 94 | ND |
| 11c | — | KHCO3 (8.0) | Acetone | 62 | 9 |
| 12d | — | KHCO3 (8.0) | Acetone | 52 | 13 |
| a Reaction conditions: 1a (0.20 mmol, 1.0 equiv.) was treated with Ag 2CO 3, Selectfluor (1.0 mmol, 5.0 equiv.), TEMPO (0.20 mmol, 1.0 equiv.), and base in organic solvent/H 2O (7.0 mL, v/v = 6/1) at 25 ℃ under argon protection for 7 h. NR = no reactions, ND = not detected. b Isolated yield. c Selectfluor (0.60 mmol, 3.0 equiv.) was used. d TEMPO (0.10 mmol, 0.5 equiv.) was used. | |||||
Silver-free decarboxylative oxygenation of uronic acids (Method A). Although decarboxylative oxygenation reaction of aliphatic carboxylic acids, especially α-aryl carboxylic acids, has been reported to yield the corresponding aldehydes or ketones under the photocatalytic [20,21] and electrochemical [22] conditions, to the best of our knowledge, such transformation of uronic acids remains unexplored. We thereby first focused our attention on optimizing the reaction conditions for decarboxylative oxygenation of uronic acids. As shown in Table 1, the control experiment indicated that KF·2H2O as a base played a decisive role in the transformation because no lactone was detected by thin-layer chromatography analysis over silica gel in the absence of this base (entry 2). These observations might be rationalized by the well-established recognition that the oxidative ability of TEMPO is highly associated with the pH value of the reaction mixture [23]. Furthermore, it was found that in the absence of Ag2CO3, exposure of 1a to the otherwise identical conditions with those in entry 2 furnished lactone 3a in 41% yield and lactol 3a' in 11% yield (entry 3), thus demonstrating that Ag2CO3 was not necessary for the transformation. Based on this finding, the reaction parameters were further scrutinized which included the types and amounts of bases, amounts of Selectfluor and TEMPO, and reaction solvents (Table 1, entries 4–12). The screenings showed that treatment of 1a with 5.0 equiv. of Selectfluor, 1.0 equiv. of TEMPO, and 8.0 equiv. of KHCO3 afforded lactone 3a in the highest yield of 94% in acetone/H2O at 25 ℃ without the help of Ag2CO3 (Table 1, entry 10). This optimized silver-free protocol was designated as Method A for decarboxylative oxygenation of uronic acids.
With the optimal conditions established, we examined the generality and limitations of Method A, and the results are shown in Scheme 2. It was found that under the action of Selectfluor and TEMPO, the decarboxylative oxygenation reaction of furanoid and pyranoid uronic acids 1b–1q proceeded smoothly, delivering the desired lactones 3b–3q in the yields ranging from 33% to 89%. These results demonstrate that the reaction enjoyed a broad substrate scope and good tolerance of functional groups. Some comments are deserved on the transformations. The combination of either TEMPO/PhI(OAc)2 or Selectfluor/NaBr is recognized oxidizing system to convert alcohols to aldehydes or ketones [24,25]. However, we found that subjection of 1b to decarboxylative oxygenation produced lactone 3b in 83% yield with the free C4-OH intact. This result underpins the feasibility of chemoselectively decarboxylative oxygenation over oxidation of a secondary hydroxy group under the current conditions. The compromised 42% yield for 3c is attributed to the formation of lactol 3c' in 32% yield due to the presence of the sterically bulky C4‑tert-butyldimethylsilyl (TBS) ether. This observation is consistent with the knowledge that the steric hindrance imposes a significant influence on the TEMPO-promoted oxidation reactions [26]. Decarboxylative oxygenation of benzoyl-protected uronic acids 1o–1q gave lactones 3o–3q in 33%–59% yields due to incomplete consumption of the starting uronic acids. Compared to the high yields of 94% and 83% obtained for 3a and 3b, the much lower yields for 3o–3q clearly illustrate the detrimental effect of benzoyl protecting groups on decarboxylative oxygenation of uronic acids when Method A was employed. These obervations highlight the limitations of this protocol to a certain extent.
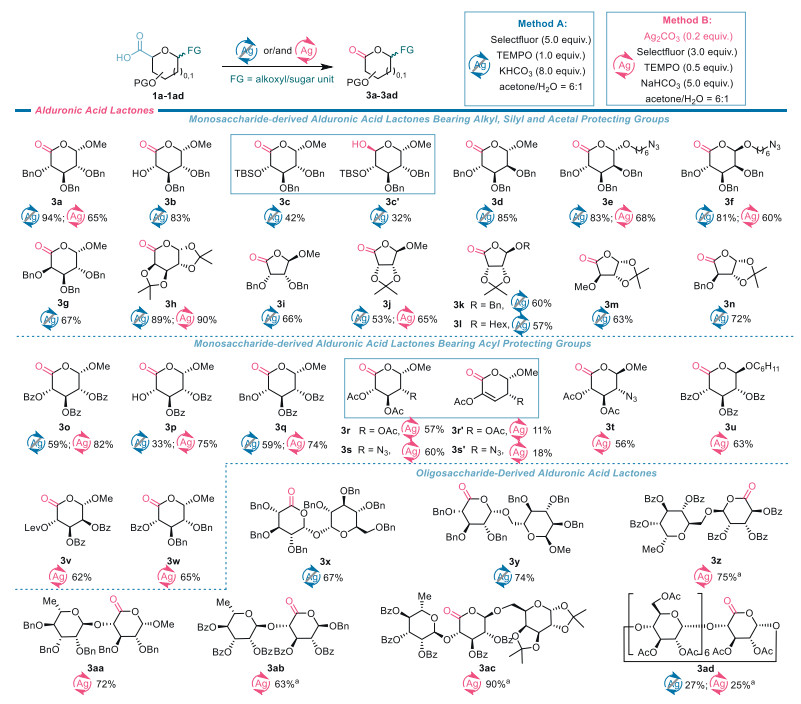
Silver-promoted decarboxylative oxygenation of uronic acids (Method B). The limitations encountered with Method A have motivated us to explore an alternative protocol for decarboxylative oxygenation in the hope of further improving the outcome of decarboxylative oxygenation of acyl-protected uronic acids. Drawing from our previous success with silver-promoted decarboxylative fluorination of acyl-protected uronic acids [17], we decided to explore the potential influence of silver salts on decarboxylative oxygenation reactions. After evaluating the reaction parameters with benzoyl-protected uronic acid 1o as a model substrate (Table S1 in Supporting information for details), we found that upon treatment with 3.0 equiv. of Selectfluor and 0.5 equiv. of TEMPO with the aid of 0.2 equiv. of Ag2CO3 in acetone/H2O at 15 ℃ for 3 h, this acid could be converted to lactone 3o in 82% yield, a much better yield than 59% obtained using Method A. This silver-assisted protocol was referred to as Method B (Scheme 2) and applied to decarboxylative oxygenation of acyl-masked uronic acids. To our satisfaction, under the conditions of Method B benzoyl-protected lactones 3p and 3q were obtained in higher yields than those obtained by Method A. It should be pointed out that the hydroxy group is also compatible with Method B as exemplified by preparation of 3p in 75% yield. Additionally, it was found that the nature of the acyl protecting groups and the configuration of the anomeric substituent exerted an influence on the outcome of silver-promoted decarboxylative oxygenation of uronic acids. Compared to benzoyl-protected glucuronic acid-based α-glycosides 1o and 1q which supplied lactones 3o and 3q exclusively, α-methyl glucoside 1r of acetyl-masked glucuronic acid gave lactone 3r in 57% yield together with α,β-unsaturated lactone 3r' in 11% yield. These observations were held by the decarboxylative oxygenation of methyl α-glycoside 1s of acetyl-protected 2-azido-2-deoxy-glucuronic acid. The reaction delivered lactone 3s and α,β-unsaturated lactone 3s' in 60% and 18% yields, respectively. We assume that 3r' and 3s' stemmed from β-elimination of the C3-OAc on 3r and 3s. In contrast to 1s, its β-anomer 1t afforded lactone 3t in 56% yield without the undesired α,β-unsaturated lactone detected. The applicability of Method B was further demonstrated by decarboxylative oxygenation of acyl-protected uronic acids 1u, 1v, and 1w that were converted into lactones 3u, 3v, and 3w in 62%–65% yields. Method B was also applicable to benzyl- and acetonide-protected uronic acids, the corresponding lactones 3a, 3e, 3f, 3h, and 3j were obtained in 60%–90% yields. At this point, comparison of the yields for compounds 3a, 3e, 3f, and 3o–3q reveals that Method A is more effective for the decarboxylative oxygenation of benzyl-protected uronic acids, while Method B is better suited for acyl-protected uronic acids.
Decarboxylative oxygenation of oligosaccharide-derived uronic acids. Either Method A or Method B for decarboxylative oxygenation was also applied to oligosaccharides (Scheme 2). Pleasingly, benzylated uronic acid 1x arising from trehalose could be transformed into lactones 3x in 67% yield using Method A. Notably, trehalose-based glycolipids are promising candidates as novel liposomal drug delivery agents [27] and as vaccine adjuvants [28] against Mycobacterium tuberculosis infections. The preparation of 3x offers a new framework for synthesizing trehalose derivatives for the related drug development. Disaccharide- and trisaccharide-derived uronic acids 1y–1ac were subjected to Method A or Method B for decarboxylative oxygenation. The reactions uneventfully provided lactones 3y–3ac in 63%–90% yields. Uronic acid 1ad prepared from β-cyclodextrin, a cyclic heptasaccharide linked by α−1,4 glucopyranosidic bonds, is also a suitable substrate. Following Method A, lactone 3ad was obtained in 27% yield. A comparable yield was obtained for decarboxylative oxygenation of 1ad by use of Method B. Notably, enzyme-catalyzed [29] and acid-mediated [30] hydrolysis reactions of β-cyclodextrin are well-established approaches to maltoheptasaccharides, availability of 3ad offers a complementary route to such oligosaccharides through the base-promoted cleavage of the lactone group.
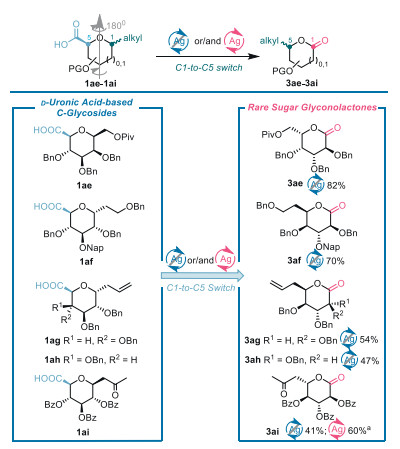
Decarboxylative oxygenation of D-uronic acid-based C-glycosides leading to rare sugar glyconolactones. Uncommon D- and L-sugar moieties are indispensable components of various natural products of biological relevance [31,32]. However, such building blocks are not easy to isolate from their natural sources. Efficient chemical protocols toward rare sugars are therefore ardently sought [31]. The C1-to-C5 switch strategy has proven to be a powerful tool for making rare sugar constructs from easily accessible D-sugar constructs [17,18,33–35].
To our delight, uronic acid-based C-glycosides are amenable to the developed decarboxylative oxygenation reactions, thus providing a novel C1-to-C5 switch approach to rare D- or L-sugar-derived glyconolactones that are otherwise difficult to make (Scheme 3). For instance, β-d-C-glycoside 1ae of mannuronic acid gave L-galactonolactone 3ae by decarboxylative oxygenation in a commendable yield of 82% using Method A. Synthesis of structurally unique 6-deoxy-d-ido-heptopyranosyl (6didoHepp) architecture is imperative but challenging for developing structurally defined glycoconjugate vaccines to combat the infections of pathogenic Campylobacter jejuni [16,33,36]. Ready access to 6-deoxy-d-ido-heptonolactone 3af and 3ag following Method A from α-d-C-glucopyranosides 1af and 1ag represents a significant advance in the synthesis of ido-heptose building blocks of interest. Moreover, the presence of the anomeric allyl group in 3ag demonstrates the tolerance of this substituent to the present reaction conditions. Additionally, Method A for decarboxylative oxygenation enabled the conversion of allyl α-d-C-allyl-galacturonic acid 1ah into the desired D-gulo-configured glyconolactone 3ah. Both 3ag and 3ah provide promising platforms for divergent construction of 6didoHepp and D-gulose settings due to rich chemistries of the lactone group and the C═C double bond. Combined with our previous work on radical decarboxylative fluorination of uronic acids of 1ag that produced oxa-[3.2.1]-bridged cycloheptane through a radical 5-exo-trig cyclization pathway [17], ready access to 3ag and 3ah with anomeric allyl group untouched further implies that a novel mechanism operates in decarboxylative oxygenation of uronic acids. Transformation of β-d-C-(2-oxo-propyl)-glucuronic acid 1ai into L-gluconolactone 3ai in 41% and 60% yields by way of Method A and Method B demonstrates the tolerance of ketone to the reaction conditions.
Mechanistic studies. Next, we moved our attention to exploring a plausible reaction mechanism with the resort to isotope-labeling experiments. By replacing H2O with H218O, decarboxylative oxygenation of uronic acid 1a was conducted under the otherwise identical conditions for either Method A or Method B (Scheme 4A). After chromatographic purification on the silica gel column, the two reactions afforded a mixture of the desired 18O-labeled lactone 18O-3a along with 3a and in the combined 83% and 65% yields, respectively. The quantitative 13C NMR spectra of the product mixture showed two signals at 169.7840 and 169.8234 ppm at an about 1:1 ratio. These two signals were assigned to the carbon of the 18O-labeled and regular carbonyl groups of the lactones, respectively. The upfield shift [Δδ(C═O)18O-16O = –5.91 Hz] of the 13C signal of the carbonyl carbon in the 18O-lactone is in agreement with those in the literature [37]. Furthermore, high-resolution mass spectroscopic analysis of the obtained mixture of 18O-3a and 3a in the mode of electron spray ionization revealed two molecular ion peaks with m/z = 468.2277 and 466.2242, corresponding to the exact molecular weight of lactone 18O-3a ([M+NH4+] calcd. for C27H32NO518O 468.2267) and that of lactone 3a ([M+NH4+] calcd. for C27H32NO6 466.2224). These results strongly suggest that water is the source of the oxygen atom in the carbonyl group of lactone in the reaction. The formation of non-labeled 3a can be attributed to the presence of unintentional water and the in situ generated water resulting from the decomposition of non-labeled NaHCO3 and Ag2CO3 in the acidic mixture. These water sources were not enriched with 18O atoms [38].
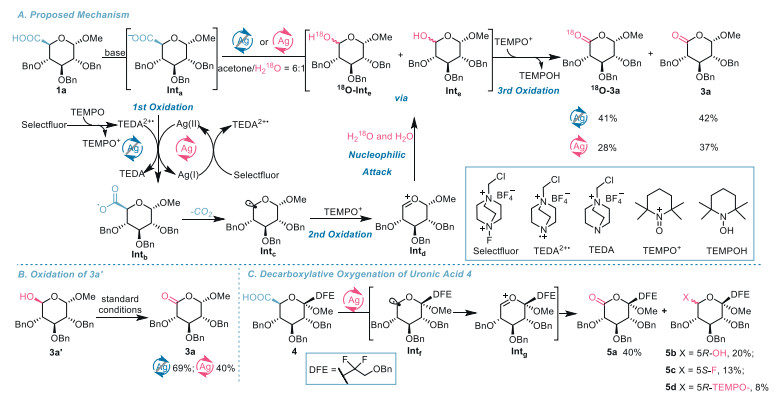
Based on these results and the precedents in the literature [39], a tentative mechanism involving an oxidative radical-polar crossover process is proposed. As shown in Scheme 4A, under the action of Selectfluor, silver(Ⅰ) species and TEMPO are oxidized to silver(Ⅱ) ions [38] and TEMPO+ respectively, along with the formation of radical cation TEDA2+• [40]. Either silver(Ⅱ) [38] or TEDA2+• [41] oxidizes the carboxylate anion Inta, generated by reacting the acid with hydrogen carbonate base, to supply acyloxy radical Intb through a single electron transfer process. Decarboxylation of Intb delivers the intermediate α-alkoxy C-centered radical Intc with extrusion of carbon dioxide. The radical species Intc is further oxidized by TEMPO+[42], affording oxocarbenium ion Intd. The attack of H2O and H218O to the carbocation ion Intd produces hemiacetals 18O-Inte and Inte which are converted into lactones 18O-3a and 3a by TEMPO+-mediated oxidation [43].
To assess the intermediacy of hemiacetal, compound 3a' was exposed to decarboxylative oxygenation under identical conditions for Method A and Method B (Scheme 4B). The reactions gave the desired lactone 3a in 69% and 40% yield, respectively, highlighting the potential of the lactol as an intermediate en route to the lactone. Additionally, besides 40% of lactone 5a and 20% of lactol 5b isolated, reverse glycosyl fluoride 5c and TEMPO-trapped product 5d were isolated in 13% and 8% yield from decarboxylative oxygenation of fluorinated uronic acid 4 (Scheme 4C). These findings are in support of the intermediacy of radical species Inte. We assume that the formation of 5c and 5d originates from either fluorine abstraction from Selectfluor or the interception of the intermediate radical Intf by TEMPO. Compared with radical species Intc, radical Intf is more challenging to oxidize to the corresponding oxacarbenium ion due to the presence of the fluorine substituent and α-oriented methoxy which lowers the reducing potential of Intf, thus offering chance for the formation of 5c and 5d in a radical manner. We ruled out the possibility of the capture of oxacarbenium ion by fluoride and TEMPOH leading to 5c and 5d because substantial excess of water as cosolvent would preferentially react with carbocation ion Intg to give the hemiacetal.
Scalable preparation and synthetic applications of xyluronic acid lactone 3a. To explore the practicality of the established methods, scale-up decarboxylative oxygenation reactions of 1a were conducted (Scheme 5). The application of both Method A and Method B yielded xyluronic acid lactone 3a on an approximately four-gram scale with yields of 91% and 64%, respectively. These results demonstrate the scalability of the developed procedures for synthesizing alduronic acid lactones and further highlight the advantage of Method A over Method B when working with benzyl-protected substrates.
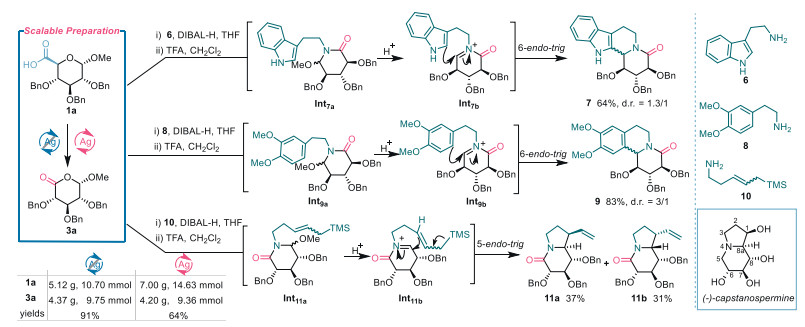
With sufficient quantities of lactone 3a in hand, we explored its divergent transformations to interesting chemotypes with natural alkaloid resemblance by coupling with the synthons having two nucleophilic sites (Scheme 5). The alkaloids with either a tetracyclic indolo[2,3-a]quinolizidine-scaffold or a tricyclic benzo[a]quinolizidine-skeleton are attractive target molecules due to their broad bioactivity profile [44]. Numerous efforts have been invested in the efficient synthesis of such compounds from pharmaceutical and organic communities [45], however, there is only one report on these alkaloids with easily accessible sugar derivatives as chirons [46]. Pleasingly, aminolysis [47] of lactone 3a with tryptamine 6 under the action of di-isobutylalumium hydride (DIBAL-H) followed by treatment with trifluoroacetic acid (TFA) smoothly provided polyhydroxylated indoloquinolizidine 7 in 64% yield with 1.3:1 diastereoselectivity. Alkaloid 7 is a structural analogue of the natural product desbromoarborescidine A [48] which exhibited antiproliferative activity. Similarly, 3a was treated with dopamine methyl ether 8 to give benzoquinolizidine 9 with d.r. 3:1 selectivity. Mechanistically, these transformations involved DIBAL-H-promoted aminolysis of lactone and the in situ formation of amido acetal (to intermediates Int7a and Int9a) as well as acid-mediated ring closure through the Pictet−Spengler reaction of N-acyliminium (intermediates Int7b and Int9b) with the aryl moieties.
Indolizidine alkaloid castanospermine has become a prospective wellspring in medicinal chemistry because of its antiviral activity against various viruses and of glycosidase inhibitory effects [49].
1-Deoxy-1-hydroxymethyl-castanospermine has been recently found to be an immunosuppressant [50]. As a consequence, many elegant strategies have been designed for the total synthesis of castanospermine and structural analogues [51]. Inspired by these precedents and to further demonstrate the synthetic versatility of alduronic acid lactones in the construction of structurally complex and bioactive alkaloids, we undertook the divergent synthesis of castanospermine-type constructs using lactone 3a and 5-(trimethylsilyl)pent‑3-en-1-amine 10 [52] as the starting materials (Scheme 5). Adopting the procedure for the synthesis of 7 and 9, the coupling reaction of 3a and 10 resulted in expeditious preparation of the indolizidine skeletons 11a and 11b as a pair of chromatographically separable diastereomers in 37% and 31% yield, respectively. The reaction involved the formation of the intermediate Int11a and cyclization of the acyliminium ion Int11b by the attack of the allyltrimethylsilyl moiety. The structures of 11a and 11b were identified by extensive 1D and 2D NMR experiments. Fortunately, a crystal of 11b was developed in MeOH and its X-ray crystallography unambiguously confirmed the structure of 11b. It should be mentioned that 11a assumes identical stereogenic centers as those in (-)-castanospermine while 11b is the 8a-epimer of 11a.
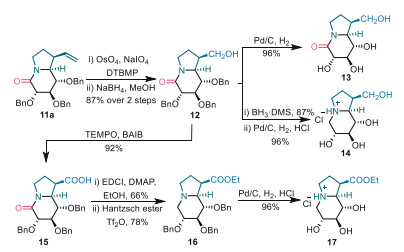
Based on rich chemistries of amido and vinyl functional groups, divergent synthesis of castanospermine-type alkaloids was then undertook by use of late-stage diversification of indolizidine frameworks 11a (Scheme 6). Oxidative cleavage of the vinyl group by NaIO4/Os4O followed by reduction of the aldehyde with NaBH4 converted 11a into primary alcohol 12 in 87% yield. Direct hydrogenolysis of the benzyl ethers in 12 with H2 over Pd/C gave lactam 13 in an excellent yield of 96%. Reduction of the tertiary amide moiety in 12 to amine with BH3·SMe2 and subsequent hydrogenolysis of the three benzyl ethers transformed 12 into indolizidine 14 in 84% overall yield. Furthermore, alcohol 12 could be oxidized to carboxylic acid 15, which was smoothly converted into ethyl ester 17 through a two-step sequence involving 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDCI)-mediated condensation with EtOH and the subsequent Pd/C-induced hydrogenolysis.
In conclusion, the radical-polar crossover decarboxylative oxygenation of uronic acids has been established as a novel and highly efficient approach to structurally diverse alduronic acid lactones and uncommon sugar glyconolactones with significant synthetic value. By utilizing Selectfluor and TEMPO as oxidants, either with or without the assistance of Ag2CO3, these transformations worked well under mild conditions, and displayed excellent compatibility with a wide variety of monosaccharide- and oligosaccharide-based uronic acids. The synthetic potential of alduronic acid lactones was elegantly demonstrated by the efficient construction of indolo[2,3-a]- or benzo[a]quinolizidine frameworks, as well as by the divergent synthesis of castanospermine-type alkaloids. Given the widespread use of alduronic acid lactones and glyconolactones as versatile chiral synthons and the frequent occurence as the common subunits in natural products, our research holds great promise for the carbohydrate-based synthesis of bioactive natural products and potential drug candidates.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Pengwei Chen: Writing – review & editing, Writing – original draft, Methodology. Xian Ma: Writing – original draft, Methodology. Ni Song: Writing – original draft, Data curation. Jianjun Wang: Resources, Data curation. Han Ding: Writing – review & editing, Formal analysis. Peng Wang: Writing – review & editing, Resources. Hongzhi Cao: Writing – review & editing. Xue-Wei Liu: Writing – review & editing. Zhihua Lv: Writing – review & editing. Ming Li: Writing – review & editing, Writing – original draft, Methodology.
We are grateful for financial support from the Marine S&T Fund of Shandong Province for Qingdao Marine Science and Technology Center (No. 2022QNLM030003-2), the National Key Research & Development Program of China (No. 2022YFA2104902), and the National Natural Science Foundation of China (Nos. 21977088 and 22377114).
Supplementary material associated with this article can be found, in the online version, at doi:
V.B. Kurteva, C.A.M. Afonso, Chem. Rev. 109 (2009) 6809–6857. doi: 10.1021/cr900169j
Q. Li, X.S. Ye, Isr. J. Chem. 55 (2015) 336–346. doi: 10.1002/ijch.201400150
D.R. Borcherding, S.A. Scholtz, R.T. Borchardt, J. Org. Chem. 52 (1987) 5457–5461. doi: 10.1021/jo00233a029
C. Hedberg, M. Estrup, E.Z. Eikeland, et al., J. Org. Chem. 83 (2018) 2154–2165. doi: 10.1021/acs.joc.7b03079
G.N. Wang, G. Reinkensmeier, S.W. Zhang, et al., J. Med. Chem. 52 (2009) 3146–3149. doi: 10.1021/jm801506m
C.G. Francisco, C.G. Martín, E. Suárez, J. Org. Chem. 63 (1998) 2099–2109. doi: 10.1021/jo971323p
J.N. Kim, E.K. Ryu, Tetrahedron Lett. 33 (1992) 3141–3144. doi: 10.1016/S0040-4039(00)79834-0
K. Tomooka, M. Kikuchi, K. Igawa, et al., Angew. Chem. Int. Ed. 39 (2000) 4502–4505. doi: 10.1002/1521-3773(20001215)39:24<4502::AID-ANIE4502>3.0.CO;2-K
M.S. Reddy, M. Narender, K.R. Rao, Tetrahedron 63 (2007) 11011–11015. doi: 10.1016/j.tet.2007.08.049
N.M. Xavier, A.P. Rauter, Y. Queneau, Carbohydrate-based lactones: synthesis and applications, in: A.P. Rauter, P. Vogel, Y. Queneau (Eds.), Carbohydrates in Sustainable Development Ⅱ, Springer, Berlin, 2010, pp. 19–62.
D. Branquet, M.V.S. Boune, N. Hucher, et al., Green Chem. 24 (2022) 7682–7688. doi: 10.1039/d2gc02606f
H. Takahashi, Y. Hitomi, Y. Iwai, et al., J. Am. Chem. Soc. 122 (2000) 2995–3000. doi: 10.1021/ja992808t
R.I. Hollingsworth, X. Song, Synlett 8 (2007) 1247–1250. doi: 10.1055/s-2007-977451
W. Dritschilo, M.K. Weibel, Biochem. Med. 9 (1974) 32–40. doi: 10.1016/0006-2944(74)90080-5
X. Zhou, H. Ding, P. Chen, et al., Angew. Chem. Int. Ed. 59 (2020) 4138–4144. doi: 10.1002/anie.201914557
Z. Hou, J. Wang, X. Zhang, et al., Chin. Chem. Lett. 34 (2023) 107804. doi: 10.1016/j.cclet.2022.107804
P. Chen, P. Wang, Q. Long, et al., Org. Lett. 22 (2020) 9325–9330. doi: 10.1021/acs.orglett.0c03514
H. Ding, N. Yan, P. Wang, et al., Org. Chem. Front. 9 (2022) 2808–2814. doi: 10.1039/d2qo00133k
E.G. Bagryanskaya, S.R.A. Marque, Chem. Rev. 114 (2014) 5011–5056. doi: 10.1021/cr4000946
S. Shirase, S. Tamaki, K. Shinohara, et al., J. Am. Chem. Soc. 142 (2020) 5668–5675. doi: 10.1021/jacs.9b12918
T.M. Faraggi, W. Li, D.W.C. MacMillan, Isr. J. Chem. 60 (2020) 410–415. doi: 10.1002/ijch.201900130
M.C. Leech, K. Lam, Acc. Chem. Res. 53 (2020) 121–134. doi: 10.1021/acs.accounts.9b00586
J.B. Gerken, Y.Q. Pang, M.B. Lauber, et al., J. Org. Chem. 83 (2018) 7323–7330. doi: 10.1021/acs.joc.7b02547
H.A. Beejapur, Q. Zhang, K. Hu, et al., ACS Catal. 9 (2019) 2777–2830. doi: 10.1021/acscatal.8b05001
H. Joshi, D. Paul, S. Sathyamoorthi, J. Org. Chem. 88 (2023) 11240–11252. doi: 10.1021/acs.joc.3c01307
M. Rafiee, K.C. Miles, S.S. Stahl, J. Am. Chem. Soc. 137 (2015) 14751–14757. doi: 10.1021/jacs.5b09672
B.S. Pattni, V.V. Chupin, V.P. Torchilin, Chem. Rev. 115 (2015) 10938–10966. doi: 10.1021/acs.chemrev.5b00046
K.T. Ryter, G. Ettenger, O.K. Rasheed, et al., J. Med. Chem. 63 (2020) 309–320. doi: 10.1021/acs.jmedchem.9b01598
H. Ji, J. Liu, D.J. McClements, et al., Crit. Rev. Food Sci. Nutr. 64 (2024) 3674–3686. doi: 10.1080/10408398.2022.2134291
A. Kikuzawa, T. Kida, M. Akashi, Org. Lett. 9 (2007) 3909–3912. doi: 10.1021/ol701449v
T.G. Frihed, M. Bols, C.M. Pedersen, Chem. Rev. 115 (2015) 3615–3676. doi: 10.1021/acs.chemrev.5b00104
Z. Guo, Y. Tang, W. Tang, et al., Nat. Prod. Rep. 38 (2021) 1887–1909. doi: 10.1039/d0np00075b
T. Li, J. Wang, X. Zhu, et al., J. Am. Chem. Soc. 143 (2021) 11171–11179. doi: 10.1021/jacs.1c05048
T.Y. Xia, Y.B. Li, Z.J. Yin, et al., Chin. Chem. Lett. 25 (2014) 1220–1224. doi: 10.1016/j.cclet.2014.06.007
Z. Qiao, D. Li, J. Gao, et al., Chin. J. Chem. 41 (2023) 3037–3044. doi: 10.1002/cjoc.202300392
P. Zhang, R. Hevey, C.C. Ling, J. Org. Chem. 82 (2017) 9662–9674. doi: 10.1021/acs.joc.7b01752
J.M. Risley, R.L.V. Etten, J. Am. Chem. Soc. 102 (1980) 4609–4614. doi: 10.1021/ja00534a007
N.R. Patel, R.A. Flowers, J. Org. Chem. 80 (2015) 5834–5841. doi: 10.1021/acs.joc.5b00826
D. Leifert, A. Studer, Chem. Rev. 123 (2023) 10302–10380. doi: 10.1021/acs.chemrev.3c00212
F.J.A. Troyano, K. Merkens, A. Gómez-Suárez, Asian J. Org. Chem. 9 (2020) 992–1007. doi: 10.1002/ajoc.202000196
F.J.A. Troyano, F. Ballaschk, M. Jaschinski, et al., Chem. Eur. J. 25 (2019) 14054–14058. doi: 10.1002/chem.201903702
A.-F. Voica, A. Mendoza, W.R. Gutekunst, et al., Nat. Chem. 4 (2012) 629–635. doi: 10.1038/nchem.1385
P. Thapa, S. Hazoor, B. Chouhan, et al., J. Org. Chem. 85 (2020) 9096–9105. doi: 10.1021/acs.joc.0c01013
P. Wu, T.E. Nielsen, Chem. Rev. 117 (2017) 7811–7856. doi: 10.1021/acs.chemrev.6b00806
M. Chrzanowska, A. Grajewska, M.D. Rozwadowska, Chem. Rev. 116 (2016) 12369–12465. doi: 10.1021/acs.chemrev.6b00315
B.V.S. Reddy, B.P. Reddy, P.V.G. Reddy, et al., Org. Biomol. Chem. 14 (2016) 4276–4282. doi: 10.1039/C6OB00250A
P.Q. Huang, X. Zheng, X.M. Deng, Tetrahedron Lett. 42 (2001) 9039–9041. doi: 10.1016/S0040-4039(01)01933-5
L. Yang, J. Li, Z. Xu, et al., Org. Lett. 24 (2022) 6531–6536. doi: 10.1021/acs.orglett.2c02466
M. Garland, S. Loscher, M. Bogyo, Chem. Rev. 117 (2017) 4422–4461. doi: 10.1021/acs.chemrev.6b00676
V.P. Vyavahare, C. Chakraborty, B. Maity, et al., J. Med. Chem. 50 (2007) 5519–5523. doi: 10.1021/jm070660f
P. Fraňová, Š. Marchalín, Eur. J. Org. Chem. 2022 (2022) e202200742. doi: 10.1002/ejoc.202200742
E. Airiau, T. Spangenberg, N. Girard, et al., Chem. Eur. J. 14 (2008) 10938–10948. doi: 10.1002/chem.200801795

Scheme 2 Synthesis of uronic acid lactones via decarboxylative oxygenation of uronic acids. Method A: uronic acids (0.2 mmol), Selectfluor (5.0 equiv.), TEMPO (1.0 equiv.), and KHCO3 (8.0 equiv.) in acetone/H2O (7.0 mL, v/v = 6/1) under argon protection at 25 ℃ for 7 h. Method B: uronic acids (0.1 mmol), Ag2CO3 (0.2 equiv.), Selectfluor (3.0 equiv.), TEMPO (0.5 equiv.), and NaHCO3 (5.0 equiv.) in acetone/H2O (3.5 mL, v/v = 6/1) under argon protection at 15 ℃ for 3 h. a Uronic acids (0.1 mmol), Ag2CO3 (0.4 equiv.), Selectfluor (6.0 equiv.), TEMPO (1.0 equiv.), and NaHCO3 (10.0 equiv.) in acetone/H2O (7 mL, v/v = 6/1).

Scheme 3 Synthesis of rare sugar glyconolactones via decarboxylative oxygenation of uronic acids. a Uronic acids (0.1 mmol), Ag2CO3 (0.4 equiv.), Selectfluor (6.0 equiv.), TEMPO (1.0 equiv.), and NaHCO3 (10.0 equiv.) in acetone/H2O (7 mL, v/v = 6/1).

Scheme 5 Scalable preparation and synthetic applications of xyluronic acid lactone 3a.
Table 1. Optimization of the reaction conditions for Method A.a
|
|||||
| Entry | Ag2CO3 (equiv.) | Base (equiv.) | Organic solvent | Yield (%)b | |
| 3a | 3a' | ||||
| 1 | 0.5 | KF·2H2O (5.0) | Acetone | 52 | 13 |
| 2 | 0.5 | — | Acetone | NR | — |
| 3 | — | KF·2H2O (5.0) | Acetone | 41 | 11 |
| 4 | — | NaHCO3 (5.0) | Acetone | 65 | 14 |
| 5 | — | NaHCO3 (5.0) | MeCN | 47 | 16 |
| 6 | — | NaHCO3 (5.0) | DMF | 7 | ND |
| 7 | — | NaHCO3 (5.0) | DMSO | NR | — |
| 8 | — | CsHCO3 (5.0) | Acetone | 80 | ND |
| 9 | — | KHCO3 (5.0) | Acetone | 83 | ND |
| 10 | — | KHCO3 (8.0) | Acetone | 94 | ND |
| 11c | — | KHCO3 (8.0) | Acetone | 62 | 9 |
| 12d | — | KHCO3 (8.0) | Acetone | 52 | 13 |
| a Reaction conditions: 1a (0.20 mmol, 1.0 equiv.) was treated with Ag 2CO 3, Selectfluor (1.0 mmol, 5.0 equiv.), TEMPO (0.20 mmol, 1.0 equiv.), and base in organic solvent/H 2O (7.0 mL, v/v = 6/1) at 25 ℃ under argon protection for 7 h. NR = no reactions, ND = not detected. b Isolated yield. c Selectfluor (0.60 mmol, 3.0 equiv.) was used. d TEMPO (0.10 mmol, 0.5 equiv.) was used. | |||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: