
Figure 1.
(a) Nanographenes containing hexa[7]circulene synthesized from [6]helicene by Scholl cyclization. (b) Study of Scholl cyclization for substituted [6]helicenes in this work.

Nanographenes with negative Gaussian curvature have garnered increasing attention in the recent decades due to their unique geometries, aromaticity as well as photophysical properties [1,2]. Most negatively curved nanographenes featured π-conjugated surfaces containing heptagonal, octagonal or even larger rings, which, together with their adjacent rings, created large steric crowdedness, ultimately rendering the π-surface non-planar [3–5]. The typical examples are [7]circulene [6,7] and its derived nanographenes [8–14], which exhibited extremely curved and twisted frameworks that deviated significantly from the planar geometry. Besides, certain nanographenes containing hexa[7]circulene (or dehydro[6]helicene) units that were formed by removing one benzene ring from the original [7]circulene also exhibited characteristic negative curvature [15–22]. These negatively curved structures possessed distinctive conformational features, leading to unique packing and assembly patterns [23,24]. However, synthesizing these negative curvatures often involves complex and laborious synthetic routes, significantly impeding the advancement. Consequently, developing novel generalized methods is revealed to be pressingly demanded.
Recently, in several nanographenes, the Scholl reaction of [6]helicene units to generate hexa[7]circulene units was revealed to be a simpler way of constructing negatively curved nanographenes (Fig. 1a) [25–27]. Such reactions could give high reactivity [28], enabling the cyclization of multiple [6]helicene units within a single nanographene [17]. Meanwhile, it is important to note that such cyclization under Scholl conditions is not always feasible [29], with the primitive carbo[6]helicene serving as a notable exception [18,26]. Therefore, a systematical investigation into the Scholl cyclization of [6]helicenes would be highly desirable [30]. In this study, we explore the Scholl cyclization of various substituted [6]helicenes, including those with methyl, methoxy, phenyl, fluoro, bromo and trifluoromethyl substituents at different positions (Fig. 1b). Our investigation reveals that the substituents on the terminal helicene rings significantly influence the reaction reactivity. Generally, electron-donating groups facilitate the Scholl cyclization, leading to the formation of hexa[7]circulene derivatives, while electron-withdrawing groups hinder the cyclization process. Furthermore, the 2,15-substituted hexa[7]circulene derivatives, owing to the steric effect caused by the introduced substituents, display stable chiral configurations against racemization, thereby emitting circularly polarized luminescence. We anticipate that this study would provide valuable insights for synthesizing negatively curved structures, thus introducing novel concepts in the design of chiral negatively curved nanographenes.
All [6]helicene precursors, including 2,15-bis-substituted and 4,13-bis-substituted derivatives, were synthesized through the photocyclization method starting from bis-vinyl-substituted naphthalenes. These precursors are designated as 6H2,15−Me (featuring methyl groups at positions 2 and 15), 6H2,15-OMe, 6H2,15-Ph, 6H2,15-Br, 6H2,15-F, 6Hnull and their analogues 6H4,13−Me, 6H4,13-OMe, 6H4,13-Ph, 6H4,13-Br, 6H4,13-F and 6Hnull. Then, we commenced our investigation into the Scholl cyclization from the less crowded 4,13-substituted [6]helicene derivatives. The reaction was conducted in CH2Cl2 solution using 1.2 equiv. of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as the oxidant, along with an excessive amount of CF3SO3H to ensure a strongly acidic environment. The reaction was taken under argon with stirring for 2 h at 25 ℃. Among the six 4,13-substituted precursors tested, 6H4,13-Me, 6H4,13-OMe and 6H4,13-Ph successfully yielded the target products H7C4,13-Me, H7C4,13-OMe and H7C4,13-Ph with the yields of 75%, 71% and 58%, respectively (Fig. 2) with removal of two hydrogen atoms according to both NMR and mass analysis. By changing the acid from CF3SO3H to CH3SO3H or CF3COOH, the Scholl reaction still gave negative results. On the contrary, 6H4,13-Br, 6H4,13-F and 6Hnull, gave no target products with employing either CF3SO3H, CH3SO3H or CF3COOH, and thin-layer chromatography (TLC) analysis indicated the formation of inseparable side-products (Tables S1 and S2 in Supporting information).
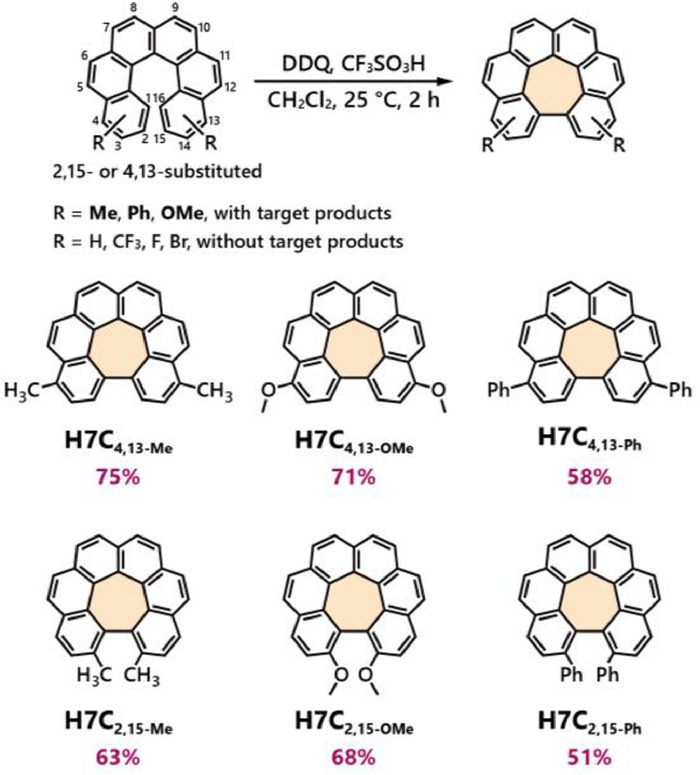
The aforementioned results suggested that substituents might significantly impact the reactivity. We hypothesized that electron-donating groups might facilitate the Scholl cyclization process, whereas electron-withdrawing groups might hinder it. To elucidate the underlying mechanism, DFT theoretical calculations were conducted. According to the Scholl mechanism, two plausible ways (the radical cation pathway and the arenium cation pathway) could be raised for this cyclization [31,32]. We firstly investigated the radical cation pathway. However, we were unable to construct the cyclized intermediates in most cases (only 6H4,13-Ph could form the cyclized intermediate), suggesting this pathway might not be suitable. Therefore, we shifted to the arenium cation pathway and successfully mapped out the reaction path for all helicenes (Figs. S63-S73 in Supporting information). Taking 6H4,13−Me as an illustrative example (Fig. 3a), the reaction commenced with a protonation at position 2 of 6H4,13−Me to form intermediate 6H4,13−Me-IM1, leading to an increase in free energy (ΔG) of 12.4 kcal/mol. Subsequently, it proceeded through a cyclization step with an activation barrier (ΔG‡) of 23.0 kcal/mol measured from the starting point, yielding the cyclized intermediate 6H4,13−Me-IM2. Following this, deprotonation and dehydrogenation reactions finally gave the product H7C4,13−Me with a total free energy release of 39.4 kcal/mol. With the change of the substituents to other electron-donating groups, helicenes 6H4,13-OMe and 6H4,13-Ph exhibited the initial protonation step with the energy increase of 12.8 and 13.5 kcal/mol, respectively, and the cyclization activation barriers were revealed to be 19.5 and 24.8 kcal/mol, respectively. For comparison, the unsubstituted 6H demonstrated similar initial protonation free energy as 13.3 kcal/mol at ortho-position or 12.0 kcal/mol at para-position, but a higher cyclization activation barrier of 25.8 or 26.8 kcal/mol. On the other hand, the electron-withdrawing groups brought higher protonation energies ranging from 16.9 to 18.5 kcal/mol, and activation barriers from 27.0 to 33.9 kcal/mol, which might be unfavorable for Scholl cyclization. These findings supported our hypothesis that electron-donating groups stabilize the cyclization transition state, thereby facilitating the Scholl cyclization.
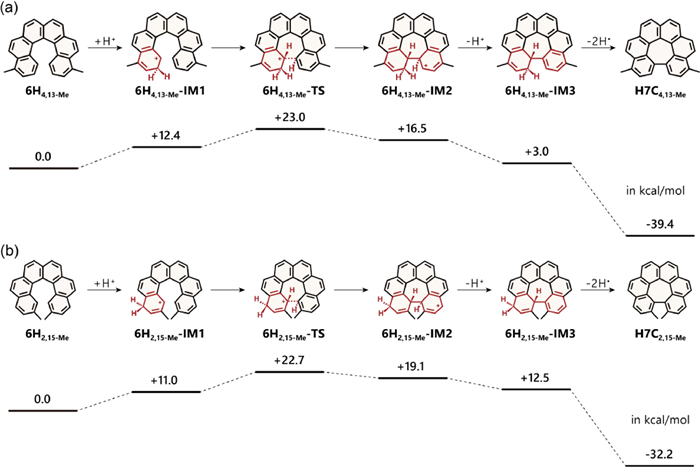
Subsequently, we investigated the 2,15-substituted [6]helicenes. Interestingly, our findings yielded similar results to those observed in 4,13-substituted species (Fig. 3b) when employing the optimized reaction condition using CF3SO3H as the acid and DDQ as the oxidant. Specifically, for methyl, methoxy and phenyl substitutions, the Scholl reaction successfully produced the desired products. However, helicenes substituted with the electron-withdrawing groups did not yield any separable products. Theoretical calculations based on the arenium cation pathway indicated that these electron-donating groups largely reduce the free energy barrier of the cyclization transition states, giving the value ranging of 19.5–22.7 kcal/mol. Conversely, the electron-withdrawing groups were found to increase the cyclization barriers in a similar manner as their 4,13-substituted homologues. It is worth noting that despite the steric crowdedness produced by the two substituents at spatially adjacent positions, the cyclization barriers were not elevated compared to the 4,13-substituted species.
By slow diffusion of n-pentane into the saturated CH2Cl2 solution for the resulting cyclized hexa[7]circulene species, we obtained the single crystals suitable for X-ray diffraction analysis for compounds H7C2,15−Me and H7C2,15-OMe (Figs. S41 and S42 in Supporting information). Both crystals showed spontaneous resolution with only one enantiomer existing in a single crystal. Taking H7C2,15-OMe for example, the structure revealed a typical negatively curved geometry, showing a saddle-like pattern (Fig. 4a). To measure the non-planarity of the π-surface, we calculated the average distance between the carbon atoms from the hexa[7]circulene unit to the averaged plane fitted by the least-squares method, giving the value of 0.63 Å.
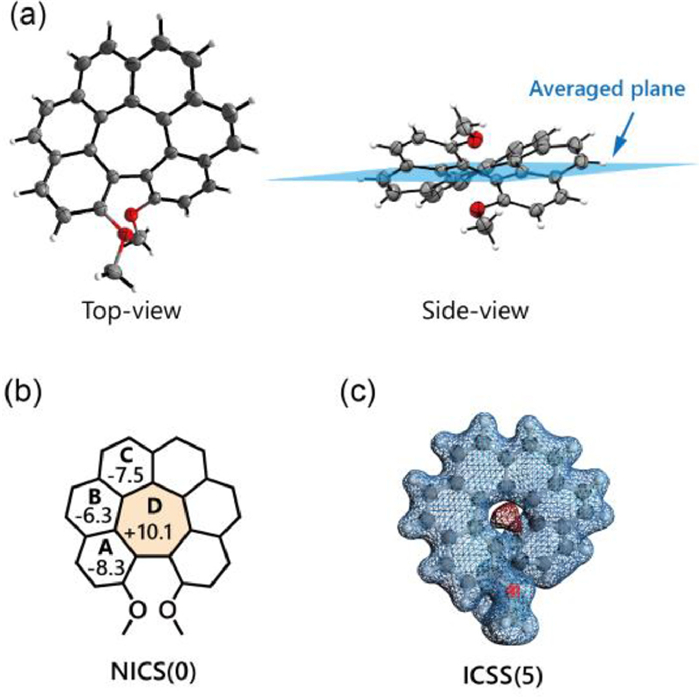
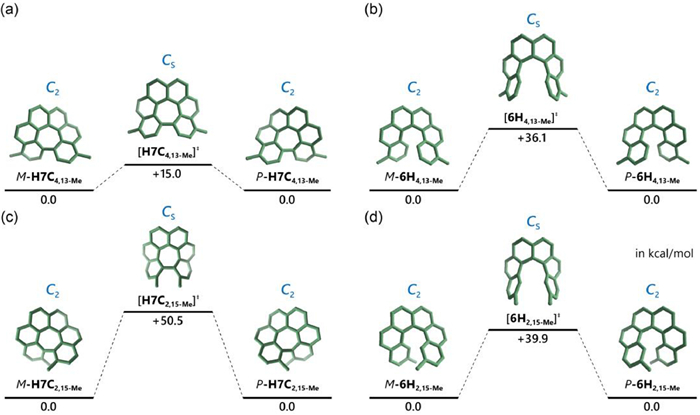
To gain a deeper understanding of the unique conformation interconversion and the chirality properties of these hexa[7]circulene species, we conducted theoretical calculations to investigate the enantiomerization process between P- and M-enantiomers. Taking H7C4,13−Me for example, the enantiomerization underwent a Cs symmetric transition state [H7C4,13−Me]‡ (Fig. 5a). This geometry could be comparable to the transition state of [6]helicene (Fig. 5b), however with a significantly lower interconversion barrier (15.0 vs. 36.1 kcal/mol) [33]. This revealed to be similar to unsubstituted hexa[7]circulene H7C (Fig. S76b in Supporting information), as well as the methoxy and phenyl substituted homologues (Figs. S77 and S79 in Supporting information), indicating that the peripheral substituents exert negligible influence on the enantiomerization barrier. Consequently, the interconversion rate k would fall within the range of 102 s−1, indicating that these molecules could not maintain their configurations stably at ambient temperature. On the contrary, substituents at positions 2 and 15 led to a notable increase in the interconversion barrier (Figs. 5c and d) [34,35]. Specifically, H7C2,15−Me demonstrated a significantly high barrier of 50.5 kcal/mol, sufficient to maintain a stable configuration at ambient temperature (Fig. 5c). Furthermore, methoxy and phenyl-substituted analogues also exhibited high values with 40.8 and 50.8 kcal/mol, respectively (Figs. S78 and S80 in Supporting information). Owing to the high interconversion barrier, we were able to successfully separate the two enantiomers of H7C2,15-Me, H7C2,15-OMe and H7C2,15-Ph using chiral HPLC. The negatively curved products we obtained exhibited intriguing aromatic characteristics. Based on the nucleus independent chemical shifts (NICS) analysis (Fig. 4b) [36,37], the central heptagonal ring (Ring D) demonstrated a positive NICS(0) value, ranging between +9.3 and +11.1 for the six molecules we synthesized (Figs. S145-S150 in Supporting information). Additionally, when compared to the uncyclized [6]helicene precursors, we observed a weakening of aromaticity in the terminal rings (Ring A) and a strengthening of aromaticity in the middle rings (Ring C). This aromaticity could also be visually represented through ICSS analysis (Fig. 4c) [38], which perceptually revealed a significant de-shielded region within the central heptagonal rings (Figs. S145-S150).
For the absorption properties, the obtained nanographenes exhibited obvious differences compared to their helicene precursors (Figs. 6a and b). Specifically, the bands of helicenes between 300 and 350 were diminished in the cases of hexa[7]circulenes, while new bands emerged in ca. 280 to 300 nm. Besides, the three 2,15-subsituted hexa[7]circulenes also displayed intense ECD signals with the two enantiomers exhibiting mirror image patterns. The absolute configuration was determined with the assistance of theoretical calculations. Notably, P-H7C2,15−Me gave a positive Cotton effect at 330 nm with Δε of +161 Lmol−1cm−1, followed by a negative Cotton effect at 284 nm with Δε of −155 Lmol−1cm−1 (Fig. 7a). According to ourcalculations, the positive Cotton effect is majorly attributed to the transitions 3 and 4 which could be ascribed to local excitation of the π-conjugated backbone based on the electron-hole analysis. Similarly, the negative Cotton effect at 284 nm predominantly contributed by transition 10, could also be ascribed to local excitation (Figs. S128, S132 and S136 in Supporting information).
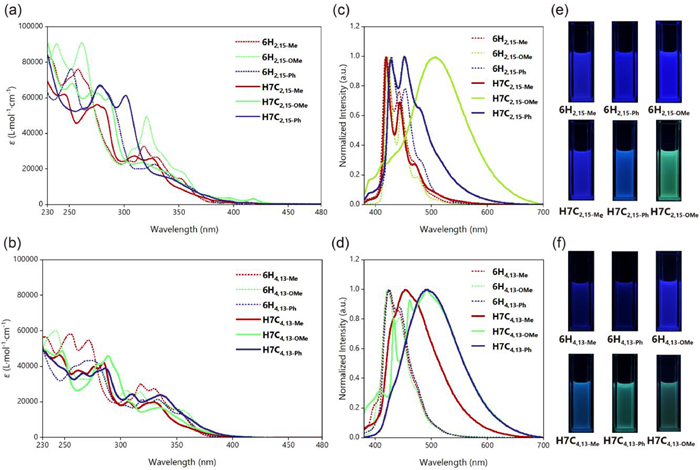
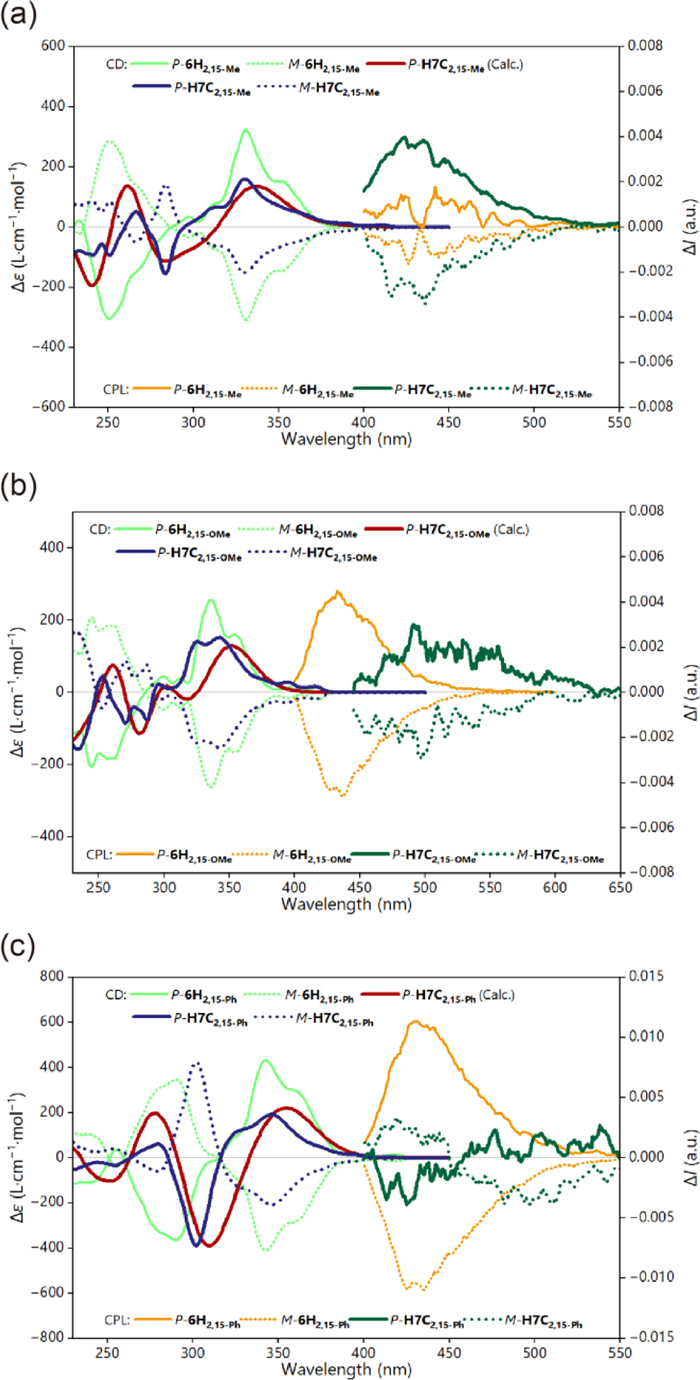
The luminescence of these hexa[7]circulenes displayed a wide range of emission colors spanning from blue to green (Figs. 6c–f). For example, the H7C2,15-OMe and H7C2,15-Ph exhibit significant red-shifted emission,while, H7C2,15−Me revealed nearly identical with 6H2,15−Me. In the case of 4,13-substituted hexa[7]circulenes, all three compounds showed red-shift compared with the corresponding [6]helicenes (Figs. 6c and d). More interestingly, we found that the emission of H7C2,15-OMe underwent a certain degree of red shift with the increase in solvent polarity (Fig. S62a in Supporting information). This diverse emission behavior contrasted significantly with their [6]helicene precursors, which solely emitted blue color. This diverse emission behavior might be largely caused by the large adjustment of the geometries in the first excited states S1 compared to the ground states S0 (Table S20 in Supporting information). Meanwhile, for helicene precursors, the geometric differences between S1 and S0 were revealed to be much less. After, we also investigated the CPL properties of the three configurationally stable hexa[7]circulenes with comparison to the corresponding [6]helicene precursors (Fig. 7). H7C2,15−Me and H7C2,15-OMe gave positive signals for P enantiomers with |glum| at ca. 4 × 10−3 and 3 × 10−3, respectively. Meanwhile, H7C2,15-Ph showed a dual signal. The CPL brightness (BCPL) (Table S5 in Supporting information) showed that H7C2,15−Me has the largest value of 1.456 Lmol−1cm−1.
In conclusion, we have conducted a comprehensive study on the cyclization behavior of a series of substituted [6]helicenes under Scholl conditions. Our findings revealed that the helicenes adorned with electron-donating groups tended to form cyclized hexa[7]circulene species whereas those analogues with electron-withdrawing groups failed to yield the desired products. Through theoretical calculations, we found that the electron-donating groups obviously reduced the activation energy barrier of the cyclization step. Furthermore, when substituted at positions 2 and 15, the obtained hexa[7]circulenes exhibited stable configurations against racemization and demonstrated circularly polarized luminescence with |glum| up to 4 × 10−3. We anticipate that this study will serve as a valuable gateway for exploring unconventional Scholl cyclization in [6]helicenes to form heptagonal rings, thereby providing novel concepts for the design and preparation of negatively curved nanographenes.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Wenxiong Yu: Writing – original draft, Methodology, Data curation. Chenyu Yang: Writing – original draft, Data curation. Xian Feng: Writing – original draft, Methodology. Chengshuo Shen: Writing – review & editing, Data curation, Conceptualization.
This work was financially supported by the Zhejiang Provincial Natural Science Foundation, China (No. LY23B040003), and the Science Foundation of Zhejiang Sci-Tech University, China (No. 22062026-Y).
Supplementary material associated with this article can be found, in the online version, at doi:
S.H. Pun, Q. Miao, Acc. Chem. Res. 51 (2018) 1630–1642. doi: 10.1021/acs.accounts.8b00140
M. Rickhaus, M. Mayor, M. Juríček, Chem. Soc. Rev. 46 (2017) 1643. doi: 10.1039/C6CS00623J
I.A.S. Chaolumen, K.E. Yamada, et al., Angew. Chem. Int. Ed. 60 (2021) 23508–23532. doi: 10.1002/anie.202100260
G.G. Miera, S. Matsubara, H. Kono, et al., Chem. Sci. 13 (2022) 1848–1868. doi: 10.1039/D1SC05586K
I.R. Márquez, S. Castro-Fernández, A. Millán, et al., Chem. Commun. 54 (2018) 6705–6718. doi: 10.1039/c8cc02325e
K. Yamamoto, T. Harada, M. Nakazaki, et al., J. Am. Chem. Soc. 105 (1983) 7171–7172. doi: 10.1021/ja00362a025
K. Yamamoto, T. Harada, Y. Okamoto, et al., J. Am. Chem. Soc. 110 (1988) 3578–3584. doi: 10.1021/ja00219a036
K. Yamamoto, Y. Saitho, D. Iwaki, T. Ooka, Angew. Chem. Int. Ed. 30 (1991) 1173–1174. doi: 10.1002/anie.199111731
K.Y. Cheung, X.M. Xu, Q. Miao, J. Am. Chem. Soc. 137 (2015) 3910–3914. doi: 10.1021/jacs.5b00403
X. Gu, H.Y. Li, B.W. Shan, Z.F. Liu, Q. Miao, Org. Lett. 19 (2017) 2246–2249. doi: 10.1021/acs.orglett.7b00714
S.H. Pun, Y.J. Wang, M. Chu, et al., J. Am. Chem. Soc. 141 (2019) 9680–9686. doi: 10.1021/jacs.9b03910
S.H. Pun, C.K. Chan, J.Y. Luo, Z.F. Liu, Q. Miao, Angew. Chem. Int. Ed. 57 (2018) 1581–1586. doi: 10.1002/anie.201711437
H.G. David, S. Míguez-Lago, C.M. Cruz, et al., Org. Mater. 03 (2021) 051–059. doi: 10.1055/s-0041-1722848
K.Y. Cheung, Q. Miao, Chin. Chem. Lett. 30 (2019) 1506–1508. doi: 10.1016/j.cclet.2019.04.013
P.J. Jessup, J.A. Reiss, Aust. J. Chem. 29 (1976) 173–178. doi: 10.1071/CH9760173
M.I. Khalid, M.S.H. Salem, S. Takizawa, Molecules 29 (2024) 296. doi: 10.3390/molecules29020296
K. Kawasumi, Q.Y. Zhang, Y. Segawa, et al., Nat. Chem. 5 (2013) 739–744. doi: 10.1038/nchem.1704
K.M. Cheung, Y.M. Xiong, S.H. Pun, et al., Chem 9 (2023) 2855–2868. doi: 10.1016/j.chempr.2023.05.028
K. Kato, H.-A. Lin, M. Kuwayama, et al., Chem. Sci. 10 (2019) 9038–9041. doi: 10.1039/c9sc03061a
K. Kato, Y. Segawa, L.T. Scott, K. Itami, Chem. Asian J. 10 (2015) 1635–1639. doi: 10.1002/asia.201500560
A. Pradhan, P. Dechambenoit, H. Bock, F. Durola, J. Org. Chem. 78 (6) (2013) 2266–2274. doi: 10.1021/jo3027752
H.A. Lin, Y. Sato, Y. Segawa, et al., Angew. Chem. Int. Ed. 57 (2018) 2874–2878. doi: 10.1002/anie.201713387
S. Zank, J.M. Fernández-García, A.J. Stasyuk, et al., Angew. Chem. Int. Ed. 61 (2022) e202112834. doi: 10.1002/anie.202112834
K. Kato, K. Takaba, S. Maki-Yonekura, et al., J. Am. Chem. Soc. 143 (2021) 5465–5469. doi: 10.1021/jacs.1c00863
J.M. Fernández-García, P.J. Evans, S.M. Rivero, et al., J. Am. Chem. Soc. 140 (2018) 17188–17196. doi: 10.1021/jacs.8b09992
C.S. Shen, G.L. Zhang, Y.L. Ding, et al., Nat. Commun. 12 (2021) 2786. doi: 10.1038/s41467-021-22992-6
Z. Qiu, S. Asako, Y. Hu, et al., J. Am. Chem. Soc. 142 (2020) 14814–14819. doi: 10.1021/jacs.0c05504
L. Zhai, R. Shukla, R. Rathore, Org. Lett. 11 (2009) 3474–3477. doi: 10.1021/ol901331p
Y. Zhang, S.H. Pun, Q. Miao, Chem. Rev. 122 (2022) 14554–14593. doi: 10.1021/acs.chemrev.2c00186
V. Akhmetov, A. Förtsch, M. Feofanov, S. Troyanovc, K. Amsharov, Org. Chem. Front. 7 (2020) 1271–1275. doi: 10.1039/d0qo00370k
P. Rempala, J. Kroulík, B.T. King, J. Org. Chem. 71 (2006) 5067–5081. doi: 10.1021/jo0526744
M. Grzybowski, K. Skonieczny, H. ButenschO $\ddot{\mathrm{n}}$, et al., Angew. Chem. Int. Ed. 52 (2013) 9900–9930. doi: 10.1002/anie.201210238
J.M. Fernández-García, P. Izquierdo-García, M. Buendía, et al., Chem. Commun. 58 (2022) 2634–2645. doi: 10.1039/d1cc06561k
C. Maeda, S. Nomoto, K. Akiyama, et al., Chem. Eur. J. 27 (2021) 15699–15705. doi: 10.1002/chem.202102269
D.Y. Yang, K.M. Cheung, Q. Gong, et al., Angew. Chem. Int. Ed. 63 (2024) e202402756. doi: 10.1002/anie.202402756
P. von Ragué Schleyer, C. Maerker, A. Dransfeld, H.J. Jiao, N.J.R. van Eikema Hommes, J. Am. Chem. Soc. 118 (1996) 6317–6318. doi: 10.1021/ja960582d
Z.F. Chen, C.S. Wannere, C. Corminboeuf, R. Puchta, P. von Ragué Schleyer, Chem. Rev. 105 (2005) 3842–3888. doi: 10.1021/cr030088+
S. Klod, E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2 (2001) 1893–1898.

Figure 1 (a) Nanographenes containing hexa[7]circulene synthesized from [6]helicene by Scholl cyclization. (b) Study of Scholl cyclization for substituted [6]helicenes in this work.

Figure 2 Scope of Scholl cyclization of a series substituted helicenes. Separation yields are listed in purple.

Figure 3 Reaction pathway for cyclization of (a) H7C4,13−Me from 6H4,13−Me and (b) H7C2,15−Me from 6H2,15−Me. Calculations were performed in the PBE0-D3(BJ)/ma-def2-TZVPP//PBE0-D3(BJ)/def-TZVP level of theory, and the Gibbs free energies are given in kcal/mol.

Figure 4 (a) Single-crystal structure of H7C2,15-OMe. (b) NICS(0) of H7C2,15-OMe. Due to the symmetry, only half of the molecules were shown. (c) ICSS analysis of H7C2,15-OMe (the numbers in parentheses refer to the absolute value of the ICSS isovalue; blue and red colors represent magnetically shielding and de-shielding areas, respectively).

Figure 5 Enantiomerization process between P- and M-isomers of (a) H7C4,13−Me, (b) 6H4,13−Me, (c) H7C2,15−Me and (d) 6H2,15−Me. Calculations were performed in the PBE0-D3(BJ)/ma-def2-TZVPP//PBE0-D3(BJ)/def-TZVP level of theory, and the Gibbs free energies are given in kcal/mol.

Figure 6 UV–vis spectra of (a) 2,15-substituted and (b) 4,13-substituted [6]helicenes and hexa[7]circulenes (c = 1 × 10–5 mol/L in CH2Cl2), fluorescence spectra of (c) 2,15-substituted and (d) 4,13-substituted [6]helicenes and hexa[7]circulenes (c = 3 × 10–5 mol/L in CH2Cl2, λex = 360 nm), and their photographs (e) and (f) under irradiation of 365 nm.

Figure 7 Experimental and calculated ECD spectra (left, c = 1 × 10–5 mol/L in CH2Cl2), and CPL spectra (right, c = 1 × 10–5 mol/L in CH2Cl2) of (a) 6H2,15−Me and H7C2,15−Me, (b) 6H2,15-OMe and H7C2,15-OMe, and (c) 6H2,15-Ph and H7C2,15-Ph. Theoretical calculations were performed at the PBE0-D3(BJ)/def-TZVP level of theory.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: